>Corresponding Author : Hassan Ala Farid
>Article Type : Case Report
>Volume : 2 | Issue : 6
>Received Date : 18 Sep, 2022
>Accepted Date : 07 Oct, 2022
>Published Date : 10 Oct, 2022
>DOI : https://doi.org/10.54289/JCRMH2200126
>Citation : Hashim AR, Farid HA, Yakob ZA, Kadhum HJ and Hasrat NH. (2022) A Family Cluster of Inherited Ataxia: Case Report and Clinical Review. J Case Rep Med Hist 2(6): doi https://doi.org/10.54289/JCRMH2200126
>Copyright : © 2022 Hashim AR, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Case Report | Open Access | Full Text
1Professor of Medicine and Neurology, College of Medicine, University of Basrah
2Post Graduate Candidate in Neurology, College of Medicine, University of Basrah
3Lecturer of Neurophysiology, College of Medicine, University of Basrah
4Assistant Professor of Medical Physiology, College of Medicine, University of Basrah
5Post Graduate Candidate in Neurophysiology, College of Medicine, University of Basrah
*Corresponding author: Hassan Ala Farid, Post Graduate Candidate in Neurology, College of Medicine, University of Basrah
Abstract
The hereditary ataxias are a highly heterogeneous group of disorders phenotypically characterised by gait ataxia, incoordination of eye movements, speech, and hand movements, and usually associated with atrophy of the cerebellum. Friedreich's ataxia is a genetic, progressive, neurodegenerative movement disorder with a typical age of onset of between 10 and 15 years. Initial symptoms may include unsteady posture, frequent falling, and progressive difficulty in walking due to an impaired ability to coordinate voluntary movements (ataxia). A family of six members was diagnosed clinically, electro-physiologically, and radiologically with having this type of inherited ataxia from Basrah, southern Iraq. This article aims to describe the clinical presentation and diagnostic approach of cerebellar ataxia, with a focus on inherited forms of ataxia.
Keywords: Ataxia; Friedreich's; Inherited Ataxia; Basrah
Abbreviations: ADEM: Acute Disseminating Encephalomyelitis, SSPE: Subacute Sclerosing Panencephalitis, ADCA: Autosomal Dominant Ataxias, DRPLA: Dentatorubral Pallidoluysian Atrophy, GSS: Gerstmann-Sträussler-Scheinker
Case presentation
A 15-year-old girl presented with progressive walking difficulties that started four years ago, associated with unsteadiness. Then the condition progressed, and she started to walk on her toe tips. She also started to develop spasticity in her upper limbs with the loss of her ability to write or perform hand skills. Recently, she started to develop chewing difficulties and slurring of speech.
She has five sisters, four of whom had similar conditions. Her father also had a similar presentation, but her mother is ultimately fit and well. Noticeably, her parents are relatives. The family pedigree is shown in Figure 1.

Figure 1: The Family Pedigree (▄ Affected male ● Affected female ⃝ Normal female)
Examination revealed circumduction gait with foot drop and high steppage gait, in addition to pes cavus and lower limb spindling. She had hypertonia (spasticity), brisk knee reflexes, and an absent ankle jerk with an equivocal plantar response. Vibration sensation and proprioception were impaired in both the upper and lower limbs. Her speech was scanning, but there was no nystagmus. Some of the clinical signs are shown in Figure 2.
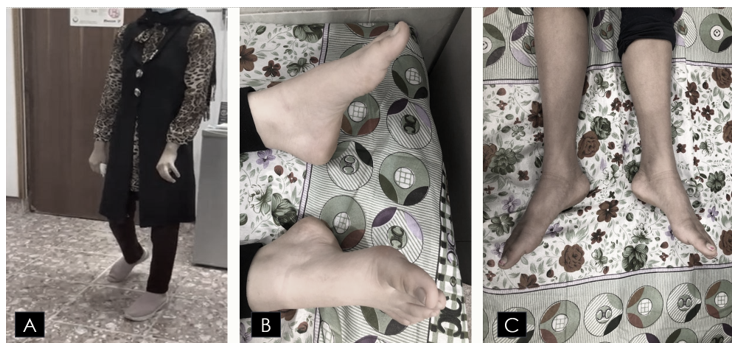
Figure 2: Some of The Clinical signs (A) demonstrates spasticity, mixed scissors and high steppage gait; (B) demonstrates bilateral pes cavus; and (C) demonstrates leg muscle wasting, spindling, and foot drop
Her investigation workup includes:
- A brain CT scan showed mild cerebellar atrophy.
- The echocardiography study was normal.
- Her random blood sugar was 187 mg/dl.
- The fundoscopic exam showed no abnormality.
- Her pure tone audiometry was also normal.
- Her spinal X-ray showed no evidence of scoliosis.
- EMG and NCS showed chronic active bilateral, symmetrical motor and sensory fibre involvement of moderate to a severe degree, affecting mainly the distal muscles of both upper and lower limbs. The primary pathology was axonal degeneration with signs of ongoing chronic denervation and chronic re-innervation.
- Her cervical spine MRI at initial presentation was normal.
According to the constellation of her symptoms, early time of onset, examination findings, which include the presence of mixed upper and lower motor neurone signs with evidence of pes cavus, family history, and investigation results, the diagnosis of inherited cerebellar ataxia is highly considered, with a high suggestion of autosomal recessive early-onset ataxia, which is most likely Friedreich ataxia. Although it is uncommon to have affected parents in autosomal recessive disorders because they are usually carriers, in this case, the father is most likely homozygous, and the mother is a carrier, so most of their children are affected by the same condition. Unfortunately, genetic testing was not available, which enables the confirmation of the mutation in the Frataxin gene.
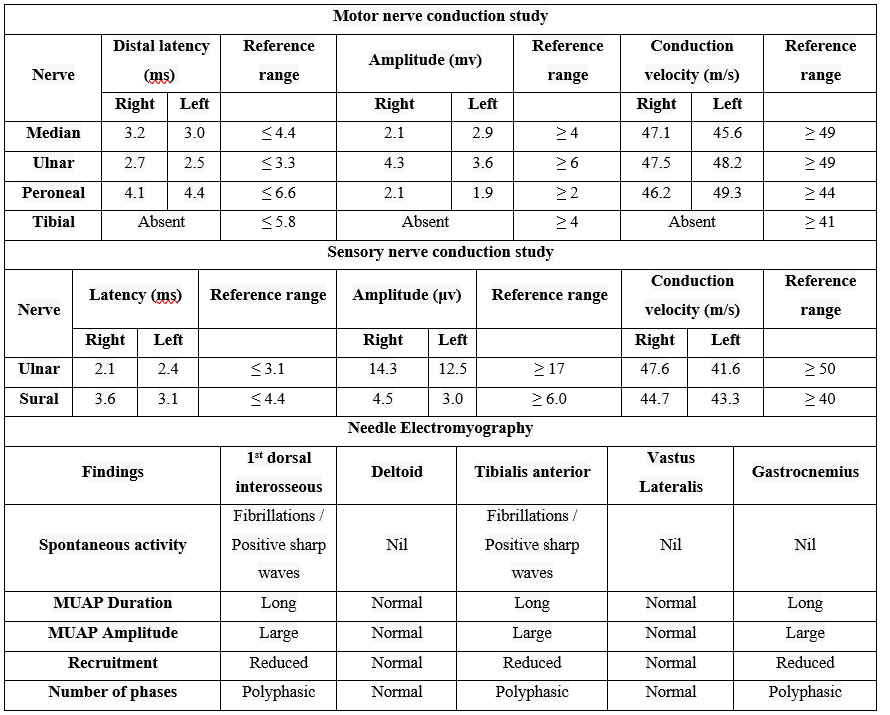
Table 1: Nerve conduction Study and EMG Parameters
Clinical Review of Cerebellar Ataxia
Ataxia implies incoordination and results from disorders of the cerebellum and its associated pathways or loss of proprioceptive sensory input in peripheral nerve disorders and spinal cord lesions affecting the posterior columns (sensory ataxia). The scope of this review is to focus on cerebellar ataxia only. The signs of cerebellar disease include [1]:
- Gait ataxia, which is described as wide-based and reeling, may be more apparent when turning or stopping suddenly.
- When mild, only tandem gait may be impaired.
- Dysmetria is an inability to perform acute finger-to-nose movements accurately with past pointing or a similar inability on heel/shin testing.
- Intention or "hunting" tremor (kinetic).
- Dysdiadochokinesia which is the inability to perform rapid alternating movements.
- Coarse nystagmus with a fast phase in the direction of the lesion; multidirectional nystagmus.
- Impaired pursuit and saccadic eye movement.
- Slurred speech and scanning dysarthria as words are broken down into syllables, with impaired volume modulation.
- Impaired heel to shin test.
- Hypotonia, hyporeflexia, and pendular reflexes.
Broadly, cerebellar ataxia can be classified into acquired and inherited ataxia. The differential diagnosis of acquired cerebellar ataxia [2]:
- Toxic substances and drugs such as alcohol, phenytoin, and metronidazole.
- Ischemic and hemorrhagic strokes.
- Multiple sclerosis, acute disseminating encephalomyelitis (ADEM), or another inflammatory (demyelination) disease.
- Neoplastic disorders, which are either metastasis from the breast and bronchus secondaries, or primary brain tumours such as pilocytic astrocytoma and medulloblastoma in children, or paraneoplastic syndrome, associated with small cell lung cancer (anti-Hu), ovarian cancer (anti-Yo), breast cancer (anti-Yo and Ri), testicular cancer (anti-Ta/Ma2), Hodgkin’s lymphoma (anti-Tr) and neuroblastoma (anti-Hu).
- Coeliac disease, gluten-related ataxia, anti-GAD-related ataxia (stiff person syndrome), or other auto-immune diseases.
- Infectious and post-infectious diseases such as viral cerebellitis (Chickenpox), measles-related subacute sclerosing panencephalitis (SSPE), and Miller Fisher syndrome (ataxia, areflexia, ophthalmoplegia + GQ1b antibody).
- Degenerative diseases such as cerebellar variants of multiple system atrophy and sporadic or variant CJD,
- Nutritional deficiencies include a lack of vitamin E and thiamine (B1).
- Structural lesions such as Arnold–Chiari malformation and arteriovenous malformation.
While the differential diagnoses of hereditary cerebellar ataxias include [3,4]:
- Autosomal dominant inheritance, which tends to present at the age of more than 25 years (late-onset) and includes the autosomal dominant ataxias (ADCAs) as well as other autosomal disorders that may have ataxia as an additional feature, such as Huntington’s disease, Dentatorubral pallidoluysian atrophy (DRPLA) and Gerstmann–Sträussler–Scheinker (GSS).
- The recessive inheritance presents at an age less than 25 (early-onset). It includes the autosomal recessive ataxias (Friedreich’s and ataxia-telangiectasia), inborn errors of metabolism, mitochondrial disorders, and episodic ataxias.
The autosomal dominant cerebellar ataxias (ADCA) include at least 50 spinocerebellar ataxia genes. Clinically, there is cerebellar ataxia in combination with any of the following: pyramidal signs (upper motor neuron), extrapyramidal signs, peripheral nerve (lower motor neuron), ophthalmoplegia, or retinopathy, and dementia. The absence of a family history does not exclude the possibility of diagnosis. There are two types of dominant ataxia [5,6]:
- ADCA type one is presented with ataxia plus pyramidal signs, ophthalmoplegia, extrapyramidal symptoms, neuropathy, and dementia. The genes involved include SCA1, SCA2, SCA3, SCA12, and SCA17.
- ADCA type two presents with ataxia, pigmentary retinopathy, and another ADCA type one symptom; the involved gene is SCA7.
- ADCA type three is presented with pure cerebellar ataxia and is usually late-onset. The involved gene is SCA6.
Autosomal recessive cerebellar ataxias (ARCA) are a heterogeneous group of rare neurological disorders involving both the central and peripheral nervous system and, in some cases, other systems and organs, and characterised by degeneration or abnormal development of the cerebellum and spinal cord, autosomal recessive inheritance, and, in most cases, early-onset occurring before the age of 20 years. The two main types of ARCA are [7]:
- Ataxia-telangiectasia, a multisystem disorder, is characterised by progressive cerebellar ataxia, ocular and cutaneous telangiectasia, and immunodeficiency with mutations in the ATM gene. Progressive ataxia develops in infancy. Telangiectasia develops later, and then patients are eventually wheelchair users and, because of associated low serum immunoglobulin levels, are susceptible to repetitive infections and malignant neoplasms (lymphoreticular tumours).
- Friedreich’s ataxia is the most common inherited ataxia. It is caused by mutations in the FRDA gene located on chromosome 9, which encodes the protein Frataxin, for which a triplet repeat expansion (GAA) is responsible. The pathology sites include degeneration, demyelination, and gliosis of the posterior columns, corticospinal tracts, dorsal spinocerebellar tracts, and ventral spinocerebellar tracts of the spinal cord, as well as dorsal roots and peripheral nerves, which are shrunken in advanced cases. Also, the cerebellum changes include Purkinje cell loss and atrophy of the dentate nucleus. In addition, peripheral nerves show loss of large, myelinated axons and segmental demyelination. The patient is usually presented with the following manifestations [8]:
o Gait ataxia.
o Pyramidal weakness with extensor planters and signs of axonal peripheral neuropathy results in features of mixed upper and lower motor neurone signs.
o Optic atrophy with abnormal eye movements, including nystagmus, broken pursuit, hypometric saccades, and macro-saccadic square-wave jerks.
o Deafness, dysarthria, and dysphagia.
o Skeletal abnormalities such as pes cavus and scoliosis.
o ECG abnormalities such as widespread T-wave inversion as well as cardiomyopathy, which are common and result in cardiac failure or dysrhythmias.
o Diabetes or glucose intolerance.
The investigations required in the cerebellar ataxia approach are guided by clinical suspicion and the possible underlying etiology. However, it is essential to perform brain imaging, send blood tests for specific antibodies as mentioned above with special consideration to celiac screen, and perform spinal imaging, electrophysiological testing, and cardiac screening to look for associated multisystemic manifestations. Nystagmography and audiometry may also be required, as well as paraneoplastic screening and molecular genetic testing [8,9].

Figure 3: Brain MRI, sagittal view, shows the evidence of cerebellar atrophy in patient with ataxia
Treatment is also dependent on the underlying etiology, as intoxications have to be cleared, vitamin deficiencies have to be replaced, endocrine disorders have to be dealt with, genetic counselling should be offered for families with inherited ataxia, and symptomatic treatment is mandatory to treat the patient suffering. Early detection and treatment of cancer are crucial; immunosuppression with corticosteroids and intravenous immunoglobulin is required in inflammatory lesions, and monitoring for the cardiac function is necessary in the case of Friedreich's ataxia [9,10].
References
- Bird TD. (2018) Hereditary Ataxia Overview. Nih.gov. [Ref.]
- Jayadev S and Bird TD. (2013) Hereditary ataxias: overview. Genetics in Medicine 15(9): 673-683. [PubMed.]
- Delatycki MB, Williamson R & Forrest SM. (2000) Friedreich ataxia: an overview. Journal of medical genetics. 37(1): 1-8. [Ref.]
- Khemani P. (2103) Overview of Adult-Onset Cerebellar Ataxia. Practical Neurology. [Ref.]
- Lindsay K, Bone I and Fuller G. (2012) Neurology and Neurosurgery Illustrated - 5th Edition. [Ref.]
- Manji H. (2014) Oxford Handbook of Neurology. blackwells.co.uk. [Ref.]
- Palau F & Espinós C. (2006) Autosomal recessive cerebellar ataxias. Orphanet journal of rare diseases. 1-47. [Ref.]
- Preston DC, Shapiro BE. (2013) Electromyography and neuromuscular disorders: clinical-electrophysiologic correlations. Elsevier Inc. Amsterdam Netherlands. [Ref.]
- Louis ED, Mayer SA and Merritt HH. (2021) Merritt’s Neurology 14th edition. [Ref.]
- de Silva RN, Vallortigara J, Greenfield J, et al. (2019) Diagnosis and management of progressive ataxia in adults. Practical Neurology. 19: 196-207. [Ref.]