>Corresponding Author : TJ Hollingsworth
>Article Type : Review Article
>Volume : 1 | Issue : 1
>Received Date : 21 September, 2021
>Accepted Date : 8 October, 2021
>Published Date : 12 October, 2021
>DOI : https://doi.org/10.54289/JORVC2100103
>Citation : Jordan KE, Jablonski MM, Hollingsworth TJ (2021) Inflammation in the Pathogenesis of Progressive Retinal Dystrophies. J Ophthalmic Res Vis Care 1(1). doi https://doi.org/10.54289/JORVC2100103
>Copyright : © 2021 Jordan KE, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Review Article | Open Access | Full Text
1Department of Ophthalmology, Hamilton Eye Institute, College of Medicine, University of Tennessee Health Science Center, Memphis, TN 38163, USA
*Corresponding author: Hollingsworth TJ, Department of Ophthalmology, Hamilton Eye Institute, College of Medicine, University of Tennessee Health Science Center, Memphis, TN 38163, USA
Abstract
Retinal degenerative diseases include inherited pathologies such as retinitis pigmentosa (RP) and multifactorial diseases such as age-related macular degeneration (AMD). AMD, a leading cause of blindness in the Western world, typically causes visual distortion and loss of central vision. In those affected, RP causes early-onset loss of night vision, followed by loss of peripheral vision and ultimately blindness. Due to the need for more effective treatments with the eventual goal of preventing blindness, researchers are investigating new potential targets and technologies to address these diseases. In a preclinical setting, various animal models are available for evaluation of investigational therapies, and investigational drugs and drug delivery methods have shown promise in both preclinical and subsequent clinical trials. Recent efforts include research into the role of inflammation in AMD and RP, and the use of anti-inflammatory drugs in preventing or even reversing retinal damage. The aim of this manuscript is to review past and current works on models for these diseases, the inflammatory processes associated with them, and potential targets for pharmacotherapy as they relate to these progressive retinal dystrophies.
Keywords: Age-related macular degeneration; retinitis pigmentosa; retinal dystrophies; inflammation; retinal degenerations; therapeutic intervention
Abbreviations: AMD: Age-related macular degeneration, RPE: Retinal Pigmented Epithelium, GA: geographic atrophy, NVAMD: neovascular AMD, CNV: choroidal neovascularization, RP: Retinitis pigmentosa, IRDs: inherited retinal dystrophies, TNFα: tumor necrosis factor alpha, NF- κB: nuclear factor-kappa B, JAK: Janus kinase, STAT: signal transducer and activator of transcription, MPGN: membranoproliferative glomerulonephritis, POS: photoreceptor outer segments, BrM: Bruch’s membrane, ONL: outer nuclear layer , RCS: Royal College of Surgeons, PDE6: Phosphodiesterase 6, VEGF: vascular endothelial growth factor, PEDF: pigment epithelium-derived factor, MMPs: matrix metalloproteinases, scFv: single chain antibody, fH: factor H, SIOP: smoke-induced ocular pathology, RIPK1: Receptor-interacting serine/threonine-protein kinase 1, RIC: RIPK1inhibitory compound, MAC: membrane attack complex, CFH: complement factor H, PAMPs: pathogen-associated molecular patterns, DAMPs: damage-associated molecular patterns, IL-1β: interleukin-1β, IL-18: interleukin-18, IL-6: interleukin-6, NLRP3: NOD-, LRR-, pyrin-domain containing protein 3, ROS: Reactive oxygen species, TLR4: toll-like receptor-4, ICAM-1: intercellular adhesion molecule-1, LFA-1: lymphocyte-associated antigen-1, LCA: Leber’s congenital amarousis, RPE65: retinal pigment epithelium-specific 65kDa protein, AAV: adeno-associated virus, UPA: upadacitinib, RAP: retinal angiomatous proliferation.
Introduction To Progressive Retinal Dystrophies
Age-related macular degeneration (AMD) is a progressive, multifactorial disease characterized by abnormalities in the retinal pigment epithelium (RPE) and photoreceptors concentrated around the macula, causing cell death with a loss of central vision. AMD is the third leading cause of irreversible blindness globally, and the leading cause of irreversible blindness in the elderly and white Americans, contributing to a high burden of disability in aging populations [1,2]. Although aging is the leading risk factor for AMD, this neurodegenerative disease is also associated with pro-inflammatory states, alterations in circadian rhythms, and excessive oxidative stress [3]. In fact, the single most crucial modifiable risk factor for AMD is smoking, a strong inducer of oxidative stress, which confers a two to three times greater risk to develop AMD compared to nonsmokers [4].
AMD can be categorized based on a few characteristic findings. Early or intermediate AMD (also called atrophic or “dry” AMD) is characterized by drusen, deposits rich in lipids and proteins that may be found between the retinal pigment epithelium and Bruch’s membrane [5]. By ophthalmoscopy, drusen appear as small, yellow deposits, which can be hard, soft, or calcific; the presence of soft drusen poses a higher risk for progression to advanced AMD and vision loss, especially if there is a high burden of large lesions [6]. Small hard drusen, on the other hand, are detectable as a normal sequalae of aging in almost all people over 50 years, and do not represent a significant risk of progression to advanced AMD [6]. Advanced AMD may refer to advanced dry AMD, characterized by geographic atrophy (GA), or it may refer to neovascular AMD (NVAMD or “wet” AMD) [5]. Nonneovascular and neovascular AMD are not mutually exclusive, and in 1-5 % of patients per year, dry AMD will progress to wet AMD [7]. Wet AMD is characterized by abnormal angiogenesis into the subretinal space, typically from the choroidal circulation, although the retinal circulation is sometimes involved [8]. These abnormal vessels can leak, causing rapid accumulation of subretinal blood and fluid [8]. While wet AMD only accounts for 10-15 % of AMD cases, it accounts for 80 % of patients who develop severe vision loss from the disease, with rapid-onset visual distortion and loss of central vision progressing over a course of days to weeks [6]. In contrast, dry AMD is characterized by gradual loss of central vision over the course of years. NVAMD can be further categorized based on patterns of choroidal neovascularization (CNV) visualized on fluorescein angiography; lesions may be classic, occult, or fibrotic, with classic lesions having a higher risk for rapid progression [9]. Retinitis pigmentosa (RP) is a group of heterogenous inherited retinal dystrophies (IRDs) caused by mutations, commonly in photoreceptor genes [10]. Nonsyndromic RP is associated with mutations in over 50 genes with variable inheritance patterns, including autosomal dominant, autosomal recessive, X-linked recessive, and mitochondrial [11]. One example of a gene product associated with both autosomal dominant and autosomal recessive RP is rhodopsin, a protein found in rod photoreceptors [11]. Rhodopsin mutations commonly cause protein misfolding and apoptosis, but mutations such as X349E and Q344X lead to mislocalization of both mutant and normal rhodopsin [12]. The defective trafficking and accumulation of rhodopsin in the inner segment, nucleus, and axons of rod photoreceptors is associated with widespread rod photoreceptor death and a more rapidly progressive clinical course of vision loss in RP [12].
While the retinopathies within this group are caused by various genetic defects, RP is typically characterized by peripheral patches of “bone-spicule” pigmentation due to the loss of photoreceptors, with rods being affected earlier than cones [10]. Clinically, these degenerative changes characteristically begin in adolescence and manifest as gradual loss of peripheral and night vision; by middle age, many patients retain only a few degrees of central vision, or may have progressed to complete blindness [11].
Inflammation, specifically proinflammatory pathways, is implicated in the pathogenesis of a wide array of retinal diseases including AMD [13] and RP [14]. These pathways include numerous cytokine-induced systems from those involving tumor necrosis factor alpha (TNFα) and nuclear factor-kappa B (NF-κB) activation to the Janus kinase/signal transducer and activator of transcription (JAK/STAT) pathway, each exerting its own detrimental effects on the retina. The purpose of this manuscript is to review past and current works on models for these diseases, the inflammatory processes associated with them, and potential targets for pharmacotherapy as they relate to these progressive retinal dystrophies.
Animal Models of AMD and RP
As mentioned previously, inflammation is known to play a role in the pathophysiology of retinal degenerative diseases. Though rodents lack a macula, they still are able to develop AMD-like phenotypes. Several rodent models aim to emulate inflammatory characteristics observed in human AMD. One example of such a model is seen in complement factor H knockout (Cfh-/-) mice. CFH usually prevents C3b from binding to complement factor B, decreasing the formation of C3 convertase. Dysregulation of this interaction in humans causes overactivation of C3, which can lead to widespread deposition. One manifestation of C3 overactivation is deposition in the kidneys, ultimately leading to membranoproliferative glomerulonephritis (MPGN) type II. The C3 deposits can also accumulate beneath the RPE, manifesting as drusen similar to that seen in AMD [16]. Similarly, Cfh-/- mice developed MPGN and retinal complement deposition, increased retinal autofluorescence, and derangement of photoreceptor outer segments (POS).
These mice also developed thinning of Bruch’s membrane (BrM), possibly due to a complement-driven increase in phagocytosis. The BrM thinning was an unexpected finding that is not characteristic of human AMD pathogenesis but may be a consequence of the opposing roles of complement in promoting both beneficial and damaging pathways in the host. For example, perhaps the uncontrolled C3 activation, while directly leading to increased complement deposition and structural subretinal damage, also leads to increased phagocytic clearance of subsequent debris [16]. In addition to the CFH knockout model, there is a transgenic mouse model with a CFH Y402H polymorphism. These mice express a chronic inflammatory phenotype due to dysregulated binding of CFH to sites of complement activation, including C-reactive protein (CRP) and heparan sulfate [16-17]. This polymorphism leads to mice with more drusen-like deposits compared to either wild-type or Cfh-/- mice. Additionally, the transgenic CFH Y402H mice had thickening of the BrM more consistent with the human model of AMD, and increased numbers of leukocytes in the subretinal space as well as increased deposits of complement in the basement membrane. Photoreceptor atrophy, while seen in Cfh-/- mice, was not observed in the CFH Y402H mice. This could be attributed to partially retained function of CFH in the transgenic mice, or it could be due to differences in age; the CFH Y402H mice were one year younger than the Cfh-/- mice when these degenerative changes were evaluated [15]. Mice transduced via an adenovirus vector to overexpress C3 exhibit loss of photoreceptor outer segments and RPE, as well as complement accumulation similar to that seen in AMD. These transgenic mice also have migration and proliferation of endothelial cells which may correspond to retinal angiomatous proliferation (RAP), a finding that has been seen in humans with NVAMD. Although these changes do provide an opportunity to study pathology relevant to both neovascular and non-neovascular AMD, this model is limited by an increased risk for retinal detachments, a finding that is not typical of AMD [15].
Cluster of differentiation 46 (CD46), also known as Membrane Cofactor Protein (MCP) is a regulatory protein that promotes inactivation of C3b and C4b. Aging Cd46-/mice exhibit signs of alternative complement pathway activation as evidenced by increased abundance of C5b-C9 in the RPE. Histologically, this model exhibits hypertrophy and vacuolization of the RPE, as well as increased autofluorescence and auto-phagocytosis in the RPE. Other phenotypic abnormalities of this model include sub-RPE deposits, thickening of BrM, reduction of choriocapillary lumen and fenestrations, and reduction of nuclei in the retinal outer nuclear layer (ONL). A model with low VEGF expression and no neovascularization, Cd46-/- mice may be a preferred option for studying non-exudative AMD in particular [17].
Another strategy in studying multifactorial diseases such as AMD has been the development of systems genetics. The BXD family of mice consists of over 150 recombinant inbred strains, originally developed from crossing C57BL/6J and DBA/2J founder mice, that has been a powerful tool in vision research [18]. The BXD family offers a relatively high diversity of phenotypes; when utilized for AMD research, combinations of haplotypes in AMD-related genes similar to those found in humans is being used to develop a more accurate mouse model of the disease. Genes that lead to altered protein function similar to the human pathophysiology can be combined, offering a unique tool to discover additional genes that may play a role in the disease, and to allow for clarification of gene-environment and gene-gene interactions that could be relevant for understanding and developing interventions of the disease [18].
Retinitis pigmentosa has been recapitulated by a variety of animal models. Royal College of Surgeons (RCS-/-) Rats is a classic model of recessively inherited retinal degeneration in which a mutation in the receptor tyrosine kinase gene Mertk results in photoreceptor death [19]. These mice display similar OCT findings compared to RP patients with autosomal recessive deletions in exon 8 of Mertk [20].
Phosphodiesterase 6 (PDE6), an enzyme involved in the phototransduction cascade, has also been manipulated to create a model of RP. For example, in mice with R560C [21] and Y347X [22] mutations in the beta subunit of PDE6, photoreceptors become locked in a depolarized state due to the lack of functional PDE6, which would usually cause closure of cGMP-gated cation channels causing hyperpolarization and slowing glutamate release at the synapse [23].
Many animal models of RP are based on mutations in the rhodopsin (RHO) gene. Rho-/- mice do not express rhodopsin, prohibiting the development of the outer segment and resulting in photoreceptor cell death [24-25]. Transgenic mice with a P23H point mutation in RHO are considered a useful model of autosomal dominant RP [26]. This point mutation results in misfolded rhodopsin and subsequent accumulation and destruction of the molecule in the endoplasmic reticulum [12, 26]. An S334X mutation in rhodopsin results in termination of the molecule without its C-terminal trafficking signal [26]. This abnormality results in mislocalization and aggregation of the mutant rhodopsin in the inner segmental, nuclear, and axonal plasma membranes; eventually, the aggregated proteins exceed the capacity of photoreceptors and trigger either caspase-dependent apoptosis or non-apoptotic cell death [12,27]. The human Q344X rhodopsin knock-in mouse, as mentioned briefly above, is associated with a rapidly progressive disease course. The Q344X mouse exhibits phenotypes expected in RP, including retinal vasculature attenuation and bone spicule pigmentation. This model also shows increased proinflammatory activity including microglial phagocytosis of viable, non-apoptotic photoreceptor cells, as well as upregulation of proinflammatory cytokines and pathways, making the Q344X mouse a possible tool in the preclinical evaluation of anti-inflammatory therapies for RP [28].
Routes of Therapeutic Administration: Bypassing Ocular Immune Privilege
The eye is shielded from deleterious effects of inflammation, in part by the blood-aqueous and blood-retina barriers [29]. This relative sequestration of the eye from the body, while providing protection of visual function, proves a challenge in the development and administration of therapeutics for ocular pathologies, especially those of the posterior segment of the eye [30]. Generally, drugs for ocular pathology may be given systemically, applied topically, intravitreally, subconjunctivally, subretinally, or suprachoroidally injected, or delivered via sustained-release implants [31]. Orally administered drugs have limited ability to access ocular tissues due to the blood-aqueous and blood-retina barriers, and as such, the doses that are required to attain a clinical advantage often cause off-target effects [32]. Topical administration is a noninvasive option that has been useful in the treatment of anterior segment pathologies but has historically not been an efficient method for drug delivery to the posterior segment of the eye [31]. A drug applied to the surface of the cornea must first permeate the corneal epithelial cells, which exhibit tight junctions between adjacent cells [33]. In addition, an ideal drug candidate must be amphipathic in order to penetrate both the lipophilic corneal epithelial layer and the hydrophilic corneal stroma [33]. One relatively new addition to our arsenal of therapeutics is the development of drug-eluting contact lenses, which increase bioavailability of drugs for anterior segment diseases by increasing the corneal contact time [34]. Historically, therapy for posterior segment diseases has mainly depended on intravitreal injections and implants. Of course, due to the more invasive nature of these compared to topical therapy, various other technologies and developments are being explored. One way in which permeability of ophthalmic solutions has been advanced is through the development of corneal penetration enhancers and drug delivery systems that enhance bioavailability. Penetration enhancers are formulations with the ability to transiently modify the structure of the epithelial cells so that drugs can enter either through the cell membranes or via a paracellular route [32]. Colloidal drug delivery systems typically consist of a penetration enhancer in addition to a bioadhesive component; this allows for increased bioavailability due to an extended drug-corneal contact time [35]. Drug delivery systems using microemulsion technology have demonstrated the ability to transport drugs with more efficacy across the corneal barrier, even when applied to drugs with inherently poor corneal permeability; innovations such as these aim to produce topical treatment options for posterior segment diseases [36].
Iontophoresis, a delivery method utilizing low intensity electric charges to briefly disrupt the ocular barrier, is another option to maximize therapy [37]. For anterior segment pathology, trans-corneal iontophoresis is effective, but for posterior segment pathology, the trans-scleral pathway offers more effective drug transportation [37].
Current Treatments for NVAMD
In NVAMD, the mainstay of treatment has been to target vascular endothelial growth factor (VEGF), a strong inducer of neovascularization [38]. Intravitreal ranibizumab is a recombinant, humanized, monoclonal antibody Fab fragment that targets VEGF-A and has been shown to prevent vision loss and improve visual acuity in patients with NVAMD [39]. Bevacizumab is another VEGF-inhibitor that, despite lacking FDA approval for the treatment of AMD, is similarly effective and far less expensive than ranibizumab as an offlabel treatment in the United States [40]. Although effective, the frequency of injections (typically once per month) needed to achieve these outcomes has proven to be a challenge for patients and clinicians [41]. One treatment that was found to allow a modest improvement in this issue is intravitreal aflibercept, which instead of requiring doses once every four weeks, could be given once every 8 weeks [42]. In an attempt to further decrease the treatment burden for patients, the Port Delivery System with ranibizumab was developed, which is an implantable, long-acting drug delivery system that was shown to have similar effects of once monthly intravitreal injections while decreasing the need for frequent treatments [43]. A more selective therapeutic option is pegaptanib, a VEGF-A165 inhibitor that spares other VEGF isoforms that may be implicated in other physiologic pathways [44]. For example, VEGF120 has been found to exert neuroprotective effects on retinal ganglion cells in the setting of ischemic injury, making a more specific VEGF inhibitor a goal of drug therapy [45]. The newest FDA-approved therapy for NVAMD is brolucizumab, a single-chain antibody fragment targeting VEGF-A that was found to be noninferior to aflibercept, but is not commonly used due to recent concerns regarding the safety [46]. Unfortunately, response to antiVEGF therapy is far from guaranteed, with anywhere from 10-22 % of patients having no improvement in symptom progression, and an additional 12 % of patients who develop tachyphylaxis to the drug class [47-48]. Therefore, additional management options need to be developed for patients with NVAMD.
Doxycycline, an antibiotic known to have anti-inflammatory effects at low doses, has been found in animal studies to have antiangiogenic effects via inhibition of M2 macrophage induction [49], as well as promotion of antiangiogenic pigment epithelium-derived factor (PEDF), resulting in reduced neovascularization [50]. Anti-inflammatory effects of doxycycline also include inhibition of hydrogen peroxideinduced oxidative stress and reduction of matrix metalloproteinases (MMPs), especially MMP-2 [51]. In a case series, patients with NVAMD were treated with oral doxycycline in addition to intraocular bevacizumab injections [52]. Patients who received oral doxycycline over the course of 4 months had similar treatment outcomes (lower incidence of intraretinal fluid or leakage, new macular hemorrhage, and reduction of visual acuity) compared to those receiving only bevacizumab with the doxycycline group requiring fewer bevacizumab injections [52]. Doxycycline is being further investigated as an MMP-9 inhibitor in patients with NVAMD who are incomplete or non-responders to intravitreal antiVEGF therapy (NCT04504123). Use of oral doxycycline in addition to traditional anti-VEGF therapy could reduce treatment burden if confirmed in larger, more robust studies.
Current Treatments for Non-Neovascular “Dry” AMD
Although approximately 90 % of patients with AMD have “dry” AMD, there are not currently any FDA approved treatments [53]. Smoking cessation and blood pressure control are standard of care and may decrease risk or rate of disease progression for patients with AMD [54]. Oral antioxidant supplements (antioxidant vitamins C, E, beta carotene, and zinc) have been considered as a preventative therapy for patients with high-risk characteristics to develop AMD, with some data supporting that these provide a decreased risk of progression to advanced AMD [54,55]. However, these supplements appear to offer more benefit in reducing progression only to exudative AMD and may have no effect on decreasing the risk of development of GA or slowing the rate of GA lesion progression [56]. Further, the use of antioxidant supplements is not without potential harm.
Genetic variation may be responsible for some inconsistencies in response to vitamin and antioxidant supplement prophylaxis, with some patients actually experiencing an increased rate of progression to NVAMD after treatment [57], while patients with vascular disease or diabetes mellitus may develop an increased risk of heart failure due to vitamin E supplementation [58]. In light of the considerable overlap of aging individuals with high risk for both AMD and cardiovascular disease, this may not be beneficial for many patients.
Investigational Therapies for Non-Neovascular “Dry” AMD
Eculizumab, a C5 inhibitor originally used in the treatment of paroxysmal nocturnal hemoglobinuria, was the first FDA approved drug to target a component of the complement system [59]. The development and success of this drug in abrogating complement pathway activation downstream of C5a and C5b has led to a gradual increase in the development of therapies against a wider array of complement pathway targets [60]. In the setting of AMD, eculizumab was evaluated in the COMPLETE study (NCT00935883), which found that intravenous infusions of eculizumab did not significantly reduce the progression of GA or reduce drusen [61]. The authors suggest that intravitreal, rather than intravenous administration of the drug, may have produced a clinical effect, as drug levels from systemic administration, while likely adequate to penetrate the choroid, are less likely to have been able to penetrate the retina or the RPE (NCT00935883).
LFG316 is an anti-C5 humanized IgG1 antibody with a modified Fc region to dampen immunogenicity [62]. A phase I clinical trial (NCT01255462) showed no significant safety concerns associated with intravitreal injection of LFG316 in patients with advanced NVAMD and/or GA. A phase II clinical trial (NCT01527500) failed to show any benefit from treatment with this drug. Another drug that has been investigated is CLG561, a fully human antibody that neutralizes properdin, a positive regulator of the alternative pathway. A subsequent phase II study (NCT02515942) investigated treatment of advanced AMD with CLG561 as a lone treatment as well as in combination with LFG316. However, this study also showed no benefit of either treatment group.
Lampalizumab, a humanized IgG anti-complement factor D antibody fragment, was the first complement-targeting therapy specifically for GA. Complement factor D is a serine protease that was targeted for the treatment of atrophic AMD due to its role in the alternative complement pathway. Lampalizumab initially showed promising results in the phase II MAHALO study (NCT02288559), where it was found to reduce GA lesion growth by 20 % in patients treated with monthly intravitreal injections compared to those receiving monthly sham injections. The MAHALO study also assessed the efficacy of lampalizumab in eyes with outer retinal tubulation (ORT) compared to eyes without ORT. The study showed that GA lesions, measured by OCT, appeared to enlarge at a slower rate in eyes with preexisting ORT compared to eyes without, implying that further studies to assess efficacy of GA may need to account for the presence of ORT [63]. However, two parallel phase III studies, SPECTRI (NCT02247531) and CHROMA (NCT02247479), were performed to further evaluate the safety and efficacy of lampalizumab in treating or slowing the progression of GA, and they were both terminated early due to lack of efficacy. Compstatins are cyclic peptides that inhibit C3 activation at the convergence point of all three complement pathways by binding to both C3 and C3b. POT-4 is a second-generation intravitreal compstatin that was evaluated for AMD treatment, and early results showed that the drug was safe and well-tolerated (NCT00473928); however, the phase II trial (NCT01603043) did not show benefit compared to sham, and there was concern for vitreal deposits of the drug secondary to administration. Subsequently, pegcetacoplan (APL-2), a reformulation of POT-4, was developed. This is a pegylated compstatin that showed promising results in the FILLY phase II trial; there was a dose dependent reduction in the progression of GA in patients treated with the drug compared to sham treatments [64]. Currently, phase III clinical trials, including DERBY (NCT03525613) and OAKS (NCT03525600), are evaluating the efficacy of intravitreal APL-2 for prevention of geographic atrophy secondary to dry AMD, with the primary outcome measure being the change in GA lesions from baseline to 12 months measured by fundus autofluorescence (FAF). Both studies anticipate a completion date of January 30, 2023.
In the GATHER1 phase 2/3 study, the anti-C5 aptamer avacincaptad pegol/ARC1905 (Zimura) also significantly reduced the growth of GA [65]. The GATHER1 study only enrolled patients with non-foveal GA secondary to AMD; the goal of excluding patients with foveal involvement was to assess the benefits of Zimura in slowing progression of the relatively more rapidly evolving lesions in sub-foveal GA patients, who were more likely to have good vision at baseline [65]. The GATHER2 phase III clinical trial is currently underway, with an estimated study completion date of July 2023 (NCT04435366).
Another potential complement-targeting treatment consists of a B4 single chain antibody (scFv) linked to a fragment of factor H (fH), an inhibitor of complement activation [66]. B4scFv-fH, expressed in microencapsulated ARPE-19 cells, has been tested in a mouse model of retinal degeneration. When injected intravitreally, the ARPE-19 cells secreted the b4scFv-fH, which resulted in decreased size of CNV lesions in a mouse model. The same drug, administered subcutaneously, also mediated reductions of retinal damage in a mouse model of smoke-induced ocular pathology (SIOP) [66]. Further, complement inhibition, in the form of the alternative pathway inhibitor CR2-fH, has been shown to not only prevent further cigarette smoke induced complement activation, but also to reverse previous smoke-induced damage in ocular disease in mouse models [67].
Corticosteroids have strong anti-inflammatory effects and inhibit activation at several distinct points in the immune system, reducing complement activation and levels of several cytokines implicated in the pathogenesis of AMD [68-70]. As such, steroids might pose an attractive therapeutic target for AMD. Iluvien is an intraocular implant that was designed to secrete fluocinolone acetate for the treatment of noninfectious posterior segment inflammatory diseases [71]. This drug is effective in the treatment of diabetic macular edema due to its inhibitory effects on many inflammatory processes including edema, fibrin deposition, capillary dilation, angiogenesis, deposition of collagen, and associated scar formation [71]. However, a phase II study (NCT00695318) to evaluate Iluvien in the treatment of GA was terminated with no apparent beneficial reduction in the GA lesions.
Doxycycline, mentioned earlier in the treatment of NVAMD, has also been evaluated for GA treatment. The TOGA study is a phase II/III study (NCT01782989); although it was completed, no results have yet been posted. Receptor-interacting serine/threonine-protein kinase 1 (RIPK1), a mediator of cell death that can be recruited by TNFα activation, is being investigated as a therapeutic target for many neurodegenerative and inflammatory diseases, including dry AMD [72]. RIPK1-inhibitory compound (RIC) was used as an ophthalmic solution in rats subsequently exposed to sodium iodate, a substance linked to the pathogenesis of dry AMD [73]. The study found that RIC was able to penetrate to the RPE and exerted a protective effect in a model of retinal degeneration [73].
Emixustat hydrochloride (ACU-4429) is a visual cycle modulator which has been evaluated in the treatment of nonexudative AMD. In animal studies, this small molecule drug has been shown to reversibly inhibit RPE65, which, in theory, could decrease visual chromophore biosynthesis and subsequent accumulation of toxic retinoid byproducts [74]. The drug had promising results in phase I and phase II studies [75-76], but it did not significantly reduce the rate of growth of GA lesions in a phase 2b/3 clinical trial [77].
Preliminary data in a small group of patients found that treatment with subcutaneous glatiramer acetate (Copoxane) has been found to reduce drusen area [78]. However, the study that supplied these results (NCT00541333) has since been suspended. Glatiramer acetate, an FDA-approved treatment for multiple sclerosis, is a drug with a poorly understood mechanism of action. In vitro studies have suggested that glatiramer acetate enhances phagocytosis, but only in a subset of monocytes [79]. The process of aging itself has been associated with dysregulated monocytes and impaired phagocytosis [80], and monocytes isolated from patients with AMD have been shown to have altered gene expression [81]. Perhaps the prophagocytic effects of glatiramer acetate may be able to compensate for immune dysregulation seen in AMD phenotypes. Additional research is needed to provide more substantial evidence for the use of this drug in AMD.
Inflammatory Targets of Non-Neovascular “Dry” AMD Therapies
In an effort to address the significant need of more effective treatments and prevention for this disabling disease, several clinical trials are evaluating therapies that target various pathways implicated in the progression of AMD, including therapies targeting inflammatory cytokines such as IL-6, IL-8, and TNFα, and targeting factors of the complement system such as C3, C5, and complement factor D [53,60,62,82]. The sources of these inflammatory cytokines are often microglia but can also be other cell types including the other glial cells of the retina (Figure 1, [84]), thus some therapies are actively working to lower microglial numbers or activity [84].
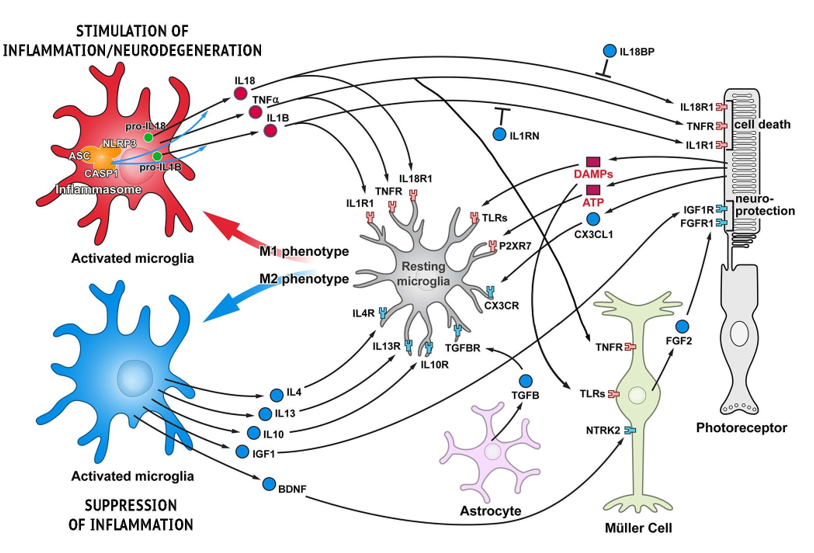
Figure 1: Functional cell-cell interactions in retina responsible for stimulating and suppressing inflammation. Schematic representation of glia-photoreceptor interactions in the retina. Three basic types of glial cell in the retina are shown: Müller cells, astroglia, and microglia. Pro-inflammatory cytokines/receptors are in red and anti-inflammatory/neuroprotective factors are marked in blue. Microglia, the resident immune cells, are responsible for initiating an inflammatory response. Microglia activation in the degenerating retina is triggered by different DAMPs, though DAMP receptors (TLRs) are also present on Müller cells allowing for proinflammatory activation in these cells as well. Although some trigger molecules which activate microglia are predicted to be released from injured cells, e.g. ATP, other molecules have not yet been identified. Activated microglia are capable of acquiring diverse phenotypes that display different cell-surface and intracellular markers, secrete different factors, and exhibit different functions. Two extreme microglial phenotypes are shown: the classically activated (M1) phenotype that promotes a pro-inflammatory response, and the alternatively activated (M2) phenotype that facilitates an anti-inflammatory response. Microglia can control their own polarization through autocrine and paracrine mechanisms. M1-polarized microglial phenotype is promoted by several cytokines, including TNF , IL-1B and IL-18. On the other hand, microglia can be driven to M2 phenotype by stimuli like IL-4, IL-13, IL-10, TGF- , and CX3CL1. Microglia, using receptors and signals, are in constant communication with neurons and other retinal cells. In pathological conditions this tight communication between cells mediate adaptive responses within the retina. Adapted from Appelbaum, Santana, and Aguirre 2017.
The complement system is a vital component of innate and adaptive immunity that protects the host from foreign pathogens. The three complement pathways are the classical, lectin, and alternative pathways [86]. The complement system plays a vital role in host homeostasis and protection through opsonization of foreign materials, production of anaphylatoxins to recruit immune cells, and the formation of a membrane attack complex (MAC) to lyse targeted cells [86]. However, chronic overactivation of the complement system has been implicated in the pathogenesis of a myriad of conditions, including autoimmune and inflammatory diseases and neurodegenerative diseases [87,88]. Complement has been demonstrated many times to be linked to the development of retinal degenerative disease, with several reports that the alternative pathway is especially implicated. A significant predisposition to develop AMD has been linked to genetic markers that are involved in the alternative pathway, such as the Y402H variant of the complement factor H (CFH) gene [89,90]. While AMD has been traditionally thought of as a disease limited to the macula, studies show evidence of systemic complement activation in these patients, which may allow for monitoring of AMD activity or progression using systemic disease markers [89].
Inflammasomes are complexes of proteins that are activated in the setting of inflammation in response to pathogenassociated molecular patterns (PAMPs) and damageassociated molecular pathogens (DAMPs). Inflammasomes promote activation of inflammatory enzymes such as caspase-1 and secretion of cytokines including interleukin-1β (IL-1β), interleukin-18 (IL-18) and interleukin-6 (IL-6) [91]. The NOD-, LRR-, and pyrin-domain containing protein 3 (NLRP3) inflammasome is an oligomeric complex that has been implicated in the pathophysiology of AMD, as well as several other inflammatory processes including host responses to viral infections, autoimmune diseases, and atherosclerosis [92].
Additionally, there have been studies to determine the significance of mitochondrial dysfunction, oxidative damage, and the accumulation of toxic metabolic by-products in the pathogenesis of AMD [62]. RPE are highly susceptible to oxidative stress, and the retina has the highest consumption of oxygen per gram of any tissue [93]. Reactive oxygen species (ROS) such as hydrogen peroxide, hydroxyl free radicals, and hydroperoxyl free radicals, are created abundantly in the environment of the RPE due its highly active metabolism as well as from exposure to UV radiation [94] . While ROS have historically been implicated in harmful effects of aging and pathophysiology of various diseases, they also are known to play a role in physiologic processes crucial to normal function. ROS are an important part of signaling pathways that allow for regulation of the innate immune response. For example, ROS are produced in response to activation of toll-like receptor-4 (TLR4) by LPS, which then leads to increased production of proinflammatory cytokines TNFα and MIP2, increased nuclear accumulation of NF-κB, and enhanced degradation of the antiinflammatory IκB-alpha [95]. ROS also induce the expression of antioxidants such as the basic leucine zipper protein NRF2, thus allowing for feedback inhibition [96].
As a normal part of aging, RPE mitochondria become less abundant and smaller in size, and the RPE cells begin to produce more waste products, such as lipofuscin [97]. In the setting of AMD, the antioxidant capacity of the retina can be overcome by excessive ROS secondary to one or more risk factors from AMD, such as smoking, a known promotor of excessive oxidative stress.
TNFα, a proinflammatory cytokine, is implicated in many inflammatory pathologies, including AMD. One of the mechanisms of TNFα is up-regulation of gene expression and subsequent activity of intercellular adhesion molecule-1 (ICAM-1), which is typically mediated through binding of
TNFα to TNF receptor-1 (TNFR1) [98]. This is accomplished via intracellular signaling pathways, in which TNFR1 complexes with TRAF2, activating downstream effectors such as PKCδ, JNK1/2, and c-Jun, eventually resulting in ICAM-1 expression [98]. ICAM-1, also known as CD54, is a cell-surface glycoprotein typically expressed on endothelial cells that has a well-known role in leukocyte transmigration as it binds to lymphocyte-associated antigen-1 (LFA-1) [99].
Through this process, TNFα leads to increased aggregation of leukocytes in various tissues, including the RPE in the setting of AMD [98].
Current Treatments for RP
Historically, patients with inherited retinal degeneration could expect to have permanent, progressive, and untreatable vision impairment. Unfortunately, development of effective treatment strategies of this group of disorders has been challenging due to the genetic, allelic, phenotypic, and clinical heterogeneity of RP [11].
While vitamin therapy and dietary modification are not effective treatment strategies for many, they do play a role in treatment for patients with one of a few rare forms of RP-like IRDs: abetalipoproteinemia (Bassen-Kornzweig syndrome), homozygous hypobetalipoproteinemia, phytanic acid oxidase deficiency (Refsum disease), and alpha-tocopherol transport protein deficiency [100,101]. Abetalipoproteinemia and hypobetalipoproteinemia, two genetic disorders characterized by impaired intestinal absorption of lipids, have an RP-like retinal degeneration as one manifestation [102]. Correction of the causative vitamin A and E deficiencies early in the disease course may slow progression of retinal degeneration More recently, investigation of various genetic targets has yielded encouraging results, with the emergence the first FDA approved gene therapy for RPE65-mediated IRD [103]. RPE65 is one of many genes implicated in autosomal recessive RP, as well as other IRDs such as Leber’s congenital amarousis (LCA) [104]. The RPE65 gene, located on chromosome 1, encodes the retinal pigment epitheliumspecific 65kDa protein (RPE65) [105]. In the classical RPEbased visual cycle, RPE65 is an isomerohydrolase, converting all-trans retinol back to the 11-cis configuration after photon absorption [105]. A lack of RPE65 leads to the accumulation of its substrates and depletion of 11-cis-retinol in the RPE [105]. In the setting of biallelic RPE65 mutations, gene replacement with subretinal injections of Voretigene Neparvovec, a recombinant adeno-associated virus (AAV) vector bearing the correct gene for RPE65 has been able to surpass this blockade of the visual cycle and improve visual function in humans [103].
In addition to gene therapy, inflammation and antiinflammatory treatments are an active field of RP research. Analyses of inflammatory gene expression and cytokine levels, especially microglia-expressed IL-1β and NLRP-3 inflammasome activation in animal models of retinal degeneration, have suggested the utility of inflammatory modulation [28,69,83]. In fact, one difference found between an early-onset model of RP and a late-onset model is that pro-inflammatory genes were actively expressed for a much longer period before the disease phenotype presented itself in the late-onset model, possibly because anti-inflammatory responses were able to compensate during this extended period [83]. These results imply that anti-inflammatory treatment could prevent, or at minimum delay, progression to blindness in those affected.
One potential anti-inflammatory therapy for RP is upadacitinib (UPA), a JAK inhibitor approved for the treatment of rheumatoid arthritis. This drug has been shown to block proinflammatory signaling in cultured retinal cells that is consistent with inflammation upregulated in RP [28]. Another study shows that dexamethasone used in a mouse model of RP caused downregulation of retinal inflammation and partial retention of retinal photoreceptor function [106]. These results imply the existence of several opportunities to evaluate existing anti-inflammatory and immune modulating therapies for the treatment of patients with RP.
Concluding Remarks
Various components of the immune system have been found to significantly contribute to the pathogenesis of retinal degenerative diseases such as AMD and RP. The abundance of existing anti-inflammatory medications, a large and varied group of therapies, is certainly an advantage of this line of investigation. Regardless of the etiology of the disease, inflammatory blockade or modulation could have substantial impact on quality of life for patients with progressive retinal disease by prolonging their sight for years, or even until more permanent therapies become available. Further evaluation of appropriate drug choices, delivery methods, and dosing regimens will be necessary to eventually secure safe, minimally invasive, and efficacious treatment options for a wide range of patients with vision-threatening retinal pathology.
References
- Wong WL, X Su, Li X, Cheung CM, Klein R, et al. (2014) Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: a systematic review and meta-analysis. Lancet Glob Health. 2(2): e106-116. [PubMed.]
- Congdon N, O’Colmain B, Klaver CCW, Klein R, Muñoz B, et al. (2004) Causes and prevalence of visual impairment among adults in the United States. Arch Ophthalmol. 122(4): 477-485. [PubMed.]
- Moles MLF, Riquelme GOL. (2016) Relationship between Oxidative Stress, Circadian Rhythms, and AMD.Oxid Med Cell Longev. 2016: 7420637. [PubMed.]
- Armstrong RA, Mousavi M. (2015) Overview of Risk Factors for Age-Related Macular Degeneration (AMD) J Stem Cells. 10(3): 171-191. [PubMed.]
- Campagne MVL, LeCouter J, Yaspan B L, Ye W. (2014) Mechanisms of age-related macular degeneration and therapeutic opportunities. J Pathol.232(2): 151-64. [PubMed.]
- Jager R D, Mieler W F, Miller J W.(2008). Age-related macular degeneration. N Engl J Med. 358(2): 2606-2617. [PubMed.]
- Bressler N M .(2004). Age-related macular degeneration is the leading cause of blindness. Jama. 291(15): 1900-1901. [PubMed.]
- Gass JDM, Agarwal A, Lavina M, Tawansy KA .(2003). Focal inner retinal hemorrhages in patients with drusen: an early sign of occult choroidal neovascularization and chorioretinal anastomosis. Retina. 23(65): 741-751. [PubMed.]
- Brown DM, Kaiser PK, Michels M, Soubrane G, Heier JS,et al.(2006). Ranibizumab versus verteporfin for neovascular age-related macular degeneration. N Engl J Med. 355(14): 1432-1444. [PubMed.]
- Hartong DT, Berson EL, and Dryja TP .(2006). Retinitis pigmentosa,.Lancet. 368(9549): 1795-1809. [PubMed.]
- Daiger SP, Sullivan LS, Bowne SJ .(2013). Genes and mutations causing retinitis pigmentosa. Clin Genet. 84(2): 132-41. [PubMed.]
- Hollingsworth TJ, Gross AK .(2012). Defective trafficking of rhodopsin and its role in retinal degenerations. Int Rev Cell Mol Biol. 293: 1-44. [PubMed.] -.(2013). The severe autosomal dominant retinitis pigmentosa rhodopsin mutant Ter349Glu mislocalizes and induces rapid rod cell death. J Biol Chem. 288(40): 29047-29055. [PubMed.]
- Kauppinen A, Paterno JJ, Blasiak J, Salminen A, Kaarniranta K.(2016). Inflammation and its role in age-related macular degeneration. Cell Mol Life Sci. 73(9): 1765-1786. [PubMed.]
- Berge JCT, Fazil Z, Born IVD, Wolfs RCW, Schreurs MWJ,et al.(2019).Intraocular cytokine profile and autoimmune reactions in retinitis pigmentosa, age-related macular degeneration, glaucoma and cataract. Acta Ophthalmol. 97(2): 185-192. [PubMed.]
- Pennesi ME, Neuringer M, Courtney RJ.( 2012). Animal models of age related macular degeneration. Mol Aspects Med. 33(4): 487-509. [PubMed.]
- Coffey PJ, Gias C, McDermott CJ, Lundh P, Pickering MC,et al.(2007). Complement factor H deficiency in aged mice causes retinal abnormalities and visual dysfunction. Proc Natl Acad Sci U S A. 104(42): 16651-16656. [PubMed.]
- Pandi SPS, Ratnayaka JA, Lotery AJ, Teeling JL.( 2021). Progress in developing rodent models of age-related macular degeneration (AMD). Exp Eye Res. 203: 108404. [PubMed.]
- Geisert EE, Williams RW.(2020). Using BXD mouse strains in vision research: A systems genetics approach. Mol Vis. 26: 173-187. [PubMed.]
- DCruz PM, Yasumura D, Weir J, Matthes MT, Abderrahim H,et al.(2000). Mutation of the receptor tyrosine kinase gene Mertk in the retinal dystrophic RCS rat. Hum Mol Genet. 9(4): 645-51. [PubMed.]
- Mackay DS, Henderson RH, Sergouniotis PI, Li Z, Moradi P, et al.(2010). Novel mutations in MERTK associated with childhood onset rod-cone dystrophy. Mol Vis. 16: 369-377. [PubMed.]
- Chang B, Hawes NL, Pardue MT, German AM, Hurd RE, et al.(2007). Two mouse retinal degenerations caused by missense mutations in the beta-subunit of rod cGMP phosphodiesterase gene. Vision Res. 47(5): 624-633. [PubMed.]
- Pittler SJ, Baehr W.(1991). Identification of a nonsense mutation in the rod photoreceptor cGMP phosphodiesterase beta-subunit gene of the rd mouse. Proc Natl Acad Sci U S A. 88(19): 8322-8326. [PubMed.]
- McLaughlin ME, Sandberg MA, Berson EL, Dryja TP .(1993). Recessive mutations in the gene encoding the beta-subunit of rod phosphodiesterase in patients with retinitis pigmentosa. Nat Genet. 4(2): 130-134. [PubMed.]
- Wang R, Jiang C, Ma J, Young MJ.(2012). Monitoring morphological changes in the retina of rhodopsin-/- mice with spectral domain optical coherence tomography,.Invest Ophthalmol Vis Sci.53(7): 3967-3972. [PubMed.]
- Gonzalez BA, Trifunović D, Sahaboglu A, Kranz K, Michalakis S.(2014). Identification of a common non-apoptotic cell death mechanism in hereditary retinal degeneration. PLoS One. 9(11): e 112142. [PubMed.]
- Nemet I, Ropelewski P, Imanishi Y .(2015). Rhodopsin Trafficking and Mistrafficking: Signals, Molecular Components, and Mechanisms. Prog Mol Biol Transl Sci. 132: 39-71. [PubMed.]
- Green ES, Menz MD, LaVail MM, Flannery JG .(2000). Characterization of rhodopsin mis-sorting and constitutive activation in a transgenic rat model of retinitis pigmentosa. Invest Ophthalmol Vis Sci. 41(16): 1546-1553. [PubMed.]
- Hollingsworth TJ, Hubbard MG, Levi HJ, White W, Wang X,et al.(2021). Proinflammatory Pathways Are Activated in the Human Q344X Rhodopsin Knock-In Mouse Model R Simpson of Retinitis Pigmentosa.Biomolecules. 11(8):1163. [PubMed.]
- Zhou R, Caspi RR .(2010). Ocular immune privilege. F1000 Biol Rep. 2:3. [PubMed.]
- Novack GD, Robin AL .(2016).Ocular pharmacology. J Clin Pharmacol. 56(5): 517-527. [PubMed.]
- Kim HM, Woo SJ .(2021). Ocular Drug Delivery to the Retina: Current Innovations and Future Perspectives. Pharmaceutics. 13(1):108. [PubMed.]
- Gaudana R, Ananthula HK, Parenky A, Mitra AK .(2010). Ocular drug delivery. Aaps j. 12(13): 348-360. [PubMed.]
- Ramsay E, Amo EMD, Toropainen E, Unadike UT, Ranta VP,et al. (2018). Corneal and conjunctival drug permeability: Systematic comparison and pharmacokinetic impact in the eye. Eur J Pharm Sci. 119: 83-89. [PubMed.]
- Phan CM, Subbaraman L, Jones L. (2014). Contact lenses for antifungal ocular drug delivery: a review. Expert opinion on drug delivery.11(4): 537-546. [PubMed.]
- Kaur IP, Smitha R (2002). Penetration enhancers and ocular bioadhesives: two new avenues for ophthalmic drug delivery. Drug Dev Ind Pharm. 28(4): 353-369. [PubMed.]
- Ibrahim MM, Maria DN, Wang X, Simpson RN, Hollingsworth TJ,et al. (2020). Enhanced Corneal Penetration of a Poorly Permeable Drug Using Bioadhesive Multiple Microemulsion Technology. Pharmaceutics. 12(8):704. [PubMed.]
- Gratieri T, Santer V, Kalia YN.(2017). Basic principles and current status of transcorneal and transscleral iontophoresis,.Expert opinion on drug delivery. 14(9): 1091-1102. [PubMed.]
- Melincovici CS, Boşca AB, Şuşman S, Mărginean M, Mihu C,et al. (2018). Vascular endothelial growth factor (VEGF) - key factor in normal and pathological angiogenesis. Rom J Morphol Embryol. 59(2): 455-467. [PubMed.]
- Rosenfeld P J, Brown DM, Heier JS, Boyer DS, Kaiser PK,et al. (2006). Ranibizumab for Neovascular Age-Related Macular Degeneration. New England Journal of Medicine.355(14): 1419-1431. [PubMed.]
- Martin DF, Maguire MG, Fine SL, Ying GS, Jaffe GJ,et al.(2020). Ranibizumab and Bevacizumab for Treatment of Neovascular Age-related Macular Degeneration: Two-Year Results. Ophthalmology. 127(4s): S135-S145. [PubMed.]
- Yancopoulos GD. (2010). Clinical application of therapies targeting VEGF. Cell. 143(1): 13-6. [PubMed.]
- Sarwar S, Clearfield E, Soliman MK, Sadiq MA, Baldwin AJ,et al. (2016). Aflibercept for neovascular age-related macular degeneration. Cochrane Database Syst Rev.2: Cd011346. [PubMed.]
- Campochiaro PA, Marcus DM, Awh CC, Regillo C, Adamis AP,et al.(2019). The Port Delivery System with Ranibizumab for Neovascular Age-Related Macular Degeneration: Results from the Randomized Phase 2 Ladder Clinical Trial. Ophthalmology. 126(8): 1141-1154. [PubMed.]
- Ng EWM, Shima DT, Calias P, Cunningham Jr ET, Guyer DR, et al.(2006). Pegaptanib, a targeted anti-VEGF aptamer for ocular vascular disease.Nat Rev Drug Discov. 5(2): 123-132. [PubMed.]
- Nishijima K, Ng YS, Zhong L, Bradley J, Schubert W,et al.(2007). Vascular endothelial growth factor-A is a survival factor for retinal neurons and a critical neuroprotectant during the adaptive response to ischemic injury. Am J Pathol. 171(1): 53-67. [PubMed.]
- Rajan K .(2020). "Update: Brolucizumabs safety under review American Academy of Ophthalmology 2020" In. [Ref.]
- Laskawiec KZ, Trzaska AK, Basta IK, Marciak WP, Dixon BR. (2019) Non-responsiveness and tachyphylaxis to anti-vascular endothelial growth factor treatment in naive patients with exudative age-related macular degeneration. J Physiol Pharmacol. 70(5). [PubMed.]
- Otsuji T, Nagai Y, Sho K, Tsumura A, Koike N,et al.(2013). Initial non-responders to ranibizumab in the treatment of age-related macular degeneration (AMD). Clin Ophthalmol. 7: 1487-1490. [PubMed.]
- He L, Marneros AG. (2014). Doxycycline inhibits polarization of macrophages to the proangiogenic M2-type and subsequent neovascularization. J Biol Chem. 289(12): 8019-8028. [PubMed.]
- Samtani S, Amaral J, Campos MM, Fariss RN, Becerra SP. (2009). Doxycycline-mediated inhibition of choroidal neovascularization. Invest Ophthalmol Vis Sci. 50(11): 5098-5106. [PubMed.]
-
Saeed M, Arun MZ, Guzeloglu M, Onursal C, Gokce G,et al.(2019). Low-dose doxycycline inhibits hydrogen peroxide-induced oxidative stress, MMP-2 up-regulation and contractile dysfunction in human saphenous vein grafts. Drug Des Devel Ther. 13: 1791-1801.
[PubMed.]
- Mirshahi A, Azimi P, Abdolahi A, Mirshahi R, Abdollahian M .(2017). Oral Doxycycline Reduces the Total Number of Intraocular Bevacizumab Injections Needed to Control Neovascular Age-related Macular Degeneration. Med Hypothesis Discov Innov Ophthalmol.6(2): 23-29. [PubMed.]
- Pytrus HMZ, Pilecka A, Kręcicka AT, Mroczek JA, Hojło MM .(2015). The Dry Form of Age-Related Macular Degeneration (AMD): The Current Concepts of Pathogenesis and Prospects for Treatment.. Adv Clin Exp Med. 24(6): 1099-1104. [PubMed.]
- Al-Zamil WM, Yassin SA .(2017). Recent developments in age-related macular degeneration: a review.Clin Interv Aging. 12: 1313-1330. [PubMed.]
- Group Age-Related Eye Disease Study Research (2001) A randomized, placebo-controlled, clinical trial of high-dose supplementation with vitamins C and E, beta carotene, and zinc for age-related macular degeneration and vision loss: AREDS report no 8. Arch Ophthalmol. 119(10): 1417-1436. [Ref.]
- Chew EY, Clemons TE, SanGiovanni JP, Danis R, Ferris RL, et al. (2013) Lutein+ zeaxanthin and omega-3 fatty acids for age-related macular degeneration: the Age-Related Eye Disease Study 2 (AREDS2) randomized clinical trial, JAMA, 309(19): 2005-2015 [Ref.]
- Vavvas DG, Small KW, Awh CC, Zanke BW, Tibshirani RJ, et al.(2018). CFH and ARMS2 genetic risk determines progression to neovascular age-related macular degeneration after antioxidant and zinc supplementation. Proc Natl Acad Sci U S A. 115(4): E696-E704. [PubMed.]
- Lonn E, Bosch J, Yusuf S, Sheridan P, Pogue J,et al.(2005). Effects of long-term vitamin E supplementation on cardiovascular events and cancer: a randomized controlled trial. Jama. 293(11): 1338-1347. [PubMed.]
- Rother RP, Rollins SA, Mojcik CF, Brodsky RA, Bell L. (2007). Discovery and development of the complement inhibitor eculizumab for the treatment of paroxysmal nocturnal hemoglobinuria. Nat Biotechnol. 25(11): 1256-1264. [PubMed.]
- Qin, S, N Dong, Yang M, Wang J, Feng X,et al.(2021). Complement Inhibitors in Age-Related Macular Degeneration: A Potential Therapeutic Option. J Immunol Res. 2021: 9945725. [PubMed.]
- Yehoshua Z, Filho CAAG, Nunes RP, Gregori G, Penha FM,et al.(2014). Systemic complement inhibition with eculizumab for geographic atrophy in age-related macular degeneration: the COMPLETE study. Ophthalmology. 121(3): 693-701. [PubMed.]
- Volz C, Pauly D .(2015). Antibody therapies and their challenges in the treatment of age-related macular degeneration. Eur J Pharm Biopharm. 95(Pt b): 158-172. [PubMed.]
- Hariri A, Nittala MG, Sadda SR .(2015). Outer retinal tubulation as a predictor of the enlargement amount of geographic atrophy in age-related macular degeneration. Ophthalmology. 122(2): 407-413. [PubMed.]
- Liao DS, Grossi FV, Mehdi DE, Gerber MR, Brown DM,et al.(2020). Complement C3 Inhibitor Pegcetacoplan for Geographic Atrophy Secondary to Age-Related Macular Degeneration: A Randomized Phase 2 Trial. Ophthalmology. 127(2): 186-195. [PubMed.]
- Jaffe GJ, Westby K, Csaky KG, Monés J, Pearlman JA,et al. (2021). C5 Inhibitor Avacincaptad Pegol for Geographic Atrophy Due to Age-Related Macular Degeneration: A Randomized Pivotal Phase 2/3 Trial. Ophthalmology. 128(4): 576-586. [PubMed.]
- Annamalai B, Parsons N, Nicholson C, Joseph K, Coughlin B, et al.(2021). Natural immunoglobulin M-based delivery of a complement alternative pathway inhibitor in mouse models of retinal degeneration. Exp Eye Res. 207: 108583. [PubMed.]
- Woodell A, Jones BW, Williamson T, Schnabolk G, Tomlinson S, et al.(2016). A Targeted Inhibitor of the Alternative Complement Pathway Accelerates Recovery From Smoke-Induced Ocular Injury. Invest Ophthalmol Vis Sci.57(4) 1728-1737. [PubMed.]
- Engelman RM, Rousou JA, Flack3rd JE, Deaton DW, Kalfin R, et al.(1995). Influence of steroids on complement and cytokine generation after cardiopulmonary bypass. Ann Thorac Surg. 60(3): 801-804. [PubMed.]
- Wooff Y, Man SM, Bruce RA, Natoli R, Fernando N. (2019). IL-1 Family Members Mediate Cell Death, Inflammation and Angiogenesis in Retinal Degenerative Diseases. Frontiers in Immunology. 10. [PubMed.]
- Tan W, Zou J, YoshidaS , Jiang B, Zhou Y .(2020). The Role of Inflammation in Age-Related Macular Degeneration. Int J Biol Sci. 16: 2989-3001. [PubMed.]
Massa H, Nagar AM, Vergados A, Dadoukis P, Patra S, et al.(2019). Intravitreal fluocinolone acetonide implant (ILUVIEN®) for diabetic macular oedema: a literature review. J Int Med Res. 47(1): 31-43. [PubMed.] - Mifflin L, Ofengeim D, Yuan J. (2020). Receptor-interacting protein kinase 1 (RIPK1) as a therapeutic target. Nature Reviews Drug Discovery. 19(8): 553-571. [PubMed.]
- Jang KH, Do YJ, Koo TS, Choi JS, Song EJ, et al.(2019). Protective effect of RIPK1-inhibitory compound in in vivo models for retinal degenerative disease. Exp Eye Res. 180: 8-17. [PubMed.]
- Bavik C, Henry SH, Zhang Y, Mitts K, McGinn T,et al. (2015). Visual Cycle Modulation as an Approach toward Preservation of Retinal Integrity. PLoS One. 10(5): e 0124940. [PubMed.]
- Kubota R, Al-Fayoumi S, Mallikaarjun S, Patil S, Bavik C, et al.(2014). Phase 1, dose-ranging study of emixustat hydrochloride (ACU-4429), a novel visual cycle modulator, in healthy volunteers. Retina. 34(3): 603-609. [PubMed.]
- Dugel PU, Novack RL, Csaky KG, Richmond PP, Birch DG,et al.(2015). Phase ii, randomized, placebo-controlled, 90-day study of emixustat hydrochloride in geographic atrophy associated with dry age-related macular degeneration. Retina. 35(6): 1173-1183. [PubMed.]
- Rosenfeld PJ, Dugel PU, Holz FG, Heier JS, Pearlman JA, et al.(2018). Emixustat Hydrochloride for Geographic Atrophy Secondary to Age-Related Macular Degeneration: A Randomized Clinical Trial. Ophthalmology. 125(10): 1556-1567. [PubMed.]
- Landa G, Butovsky O, Shoshani J, Schwartz M, Pollack A .(2008). Weekly vaccination with Copaxone (glatiramer acetate) as a potential therapy for dry age-related macular degeneration. Curr Eye Res.33(11): 1011-1013. [PubMed.]
- Gu BJ, Huang X, Avula PK, Caruso E, Drysdale C, et al.(2021). Deficits in Monocyte Function in Age Related Macular Degeneration: A Novel Systemic Change Associated With the Disease. Front Med (Lausanne). 8: 634177. [PubMed.]
- Hearps AC, Martin GE, Angelovich TA, Cheng WJ, Maisa A, et al.(2012). Aging is associated with chronic innate immune activation and dysregulation of monocyte phenotype and function .Aging Cell.11(5): 867-75. [PubMed.]
- Gruni M, Levi SH, Rinsky B, Smith Y, Chowers I .(2016). Transcriptome Analysis on Monocytes from Patients with Neovascular Age-Related Macular Degeneration .Sci Rep. 6: 29046. [PubMed.]
- Johnson LV, Leitner WP, Staples MK, Anderson DH .(2001). Complement activation and inflammatory processes in Drusen formation and age related macular degeneration. Exp Eye Res. 73(6): 887-896. [PubMed.]
- Appelbaum T, Santana E, Aguirre GD. (2017 ).Strong upregulation of inflammatory genes accompanies photoreceptor demise in canine models of retinal degeneration. PLoS One. 12(5): e0177224. [PubMed.]
- Rashid K, Schaefer IA, Langmann T .(2019) Microglia in Retinal Degeneration,.Front Immunol. 10: 1975. [PubMed.]
- Eberhard, HJM. (1988). Molecular organization and function of the complement system 1988. Ann Rev Biochem: 57.321-47. [PubMed.]
- Reis, ES, Mastellos DC, Hajishengalli G , Lambris JD .(2019). New insights into the immune functions of complement. Nat Rev Immunol. 19(8): 503-516. [PubMed.]
- Wu T, Dejanovic B, Gandham VD, Gogineni A, Edmonds R, et al.(2019). Complement C3 Is Activated in Human AD Brain and Is Required for Neurodegeneration in Mouse Models of Amyloidosis and Tauopathy. Cell Re. 28(8): 2111-23e6. [PubMed.]
- Vignesh P, Rawat A, Sharma M, Singh S .(2017). Complement in autoimmune diseases. Clin Chim Acta. 465: 123-130. [PubMed.]
- Scholl HPN, Issa PC, Walier M, Janzer S, Kopp BP, et al.(2008). Systemic complement activation in age-related macular degeneration.PLoS One.3(7): e2593. [PubMed.]
- Toomey CB, Johnson LV, s Rickman C B.(2018). Complement factor H in AMD: Bridging genetic associations and pathobiology.Prog Retin Eye Res. 62: 38-57. [PubMed.]
- Zhang W J, Chen SJ, Zhou SC, Wu SZ, Wang S.(2021). Inflammasomes and Fibrosi.Front Immunol. 12: 643149. [PubMed.]
- Zhao C, Zhao W .(2020). NLRP3 Inflammasome-A Key Player in Antiviral Responses. Front Immunol. 11: 211. [PubMed.]
- Schmidt M, Giessl A, Laufs T, Hankeln T, Wolfrum U, et al. (2003) How does the eye breathe? Evidence for neuroglobin-mediated oxygen supply in the mammalian retina. J Biol Chem. 278(3): 1932-1935. [PubMed.] Halliwell B. (1991) Reactive oxygen species in living systems: source, biochemistry, and role in human disease. Am J Med. 91(3C): 14s-22s. [PubMed.]
- Mitra S and Abraham E (2006) Participation of superoxide in neutrophil activation and cytokine production. Biochim Biophys Acta. 1762(8): 732-741. [PubMed.]
- Nagai N, Thimmulappa RK, Cano M, Fujihara M, Izumi-Nagai K, et al. (2009) Nrf2 is a critical modulator of the innate immune response in a model of uveitis. Free Radic Biol Med. 4793): 300-306. [Ref.]
- Delori FC, Goger DG, Dorey CK. (2001) Age-related accumulation and spatial distribution of lipofuscin in RPE of normal subjects. Invest Ophthalmol Vis Sci. 42(8): 1855-1866. [PubMed.]
- Lee IT, Liu SW, Chi PL, Lin CC, Hsiao LD, et al. (2015) TNF-α mediates PKCδ/JNK1/2/c-Jun-dependent monocyte adhesion via ICAM-1 induction in human retinal pigment epithelial cells. PLoS One. 10: e0117911. [Ref.]
- Verma NK, Kelleher D. (2019) An Introduction to LFA-1/ICAM-1 Interactions in T-Cell Motility. Methods Mol Biol. 1930: 1-9. [PubMed.]
- Chowers I, Banin E, Merin S, Cooper M, Granot E. (2001) Long-term assessment of combined vitamin A and E treatment for the prevention of retinal degeneration in abetalipoproteinaemia and hypobetalipoproteinaemia patients. Eye (Lond). 15(4): 525-530. [PubMed.]
- Burnett J R, Hooper AJ. (2015) Vitamin E and oxidative stress in abetalipoproteinemia and familial hypobetalipoproteinemia. Free Radic Biol Med. 88(A): 59-62. [PubMed.]
- Lee J, Hegele RA. (2014) Abetalipoproteinemia and homozygous hypobetalipoproteinemia: a framework for diagnosis and management. J Inherit Metab Dis. 37: 333-339. [PubMed.]
- Russell S, Bennett J, Wellman JA, Chung DC, Yu ZF, et al. (2017) Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet. 390(10097): 849-860. [PubMed.]
- Aoun M, Passerini I, Chiurazzi P, Karali M, De Rienzo I, et al. (2021) Inherited Retinal Diseases Due to RPE65 Variants: From Genetic Diagnostic Management to Therapy. Int J Mol Sci. 22(13):7207. [PubMed.]
- Cai X, Conley SM, Naash MI. (2009) RPE65: role in the visual cycle, human retinal disease, and gene therapy. Ophthalmic Genet. 30(2): 57-62. [PubMed.]
- Guadagni V, Biagioni M, Novelli E, Aretini P, Mazzanti CM, et al. (2019) Rescuing cones and daylight vision in retinitis pigmentosa mice. Faseb j. 33(9): 10177-92. [PubMed.]