>Corresponding Author : Mark E Peacock
>Article Type : Case Report
>Volume : 6 | Issue : 1
>Received Date : 10 Feb, 2026
>Accepted Date : 20 Feb, 2026
>Published Date : 24 Feb, 2026
>DOI : https://doi.org/10.54289/JDOE2600101
>Citation : Timothius CJC, Stern I, Stern JK, Abdelsayed RA, Ghaly M, et al. (2026) Pachyonychia Congenita: Clinical and Pathological Case Report. J Dent Oral Epidemiol 6(1): doi https://doi.org/10.54289/JDOE2600101
>Copyright : © 2026 Timothius CJC, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Case Report | Open Access | Full Text
1Department of Periodontics, Dental College of Georgia at Augusta University, Augusta, Georgia
2Department of Oral Biology and Diagnostic Sciences, Dental College of Georgia at Augusta University, Augusta, Georgia
*Corresponding author: Mark E Peacock, Department of Periodontics, Dental College of Georgia at Augusta University, Augusta, Georgia
Abstract
Introduction: Pachyonychia congenita (PC) is a very rare genetic condition triggered by mutations in one of five genes that encode for keratin. It is usually characterized by nail dystrophy and painful palmoplantar keratomas, as well as oral leukokeratosis, natal/prenatal teeth, and sebaceous cysts. This case details PC in an adult, to include the management of the patient and clinicopathologic characteristics of the disorder.
Case Description: A 78-year-old male patient with a known history of pachyonychia congenita presented with clinical findings of xerostomia, coated tongue, abnormal bilateral white lesions, and hyper-keratinization on the lateral borders of the tongue and buccal mucosa. Biopsy was obtained and pathological investigation was performed to rule out pre-cancerous lesions or other pathological conditions that may be related to the patient’s symptoms.
Practical Implications: This report describes the management of PC in an older adult patient, and reviews pertinent literature on the lesion. There is no cure for this chronic disorder, and treatment is focused on alleviating painful symptoms. Improving the quality of life is imperative for patients with this debilitating disease.
Keywords: Pachyonychia Congenita; Oral Leukokeratosis; Ectodermal Dysplasia
Abbreviations: PC: Pachyonychia congenita
Summary
This case documents a rare genetic disorder which can lead to multiple systemic and oral health problems, and includes the management of the patient and clinicopathologic characteristics of the disorder.
Introduction
Pachyonychia congenita (PC) is an exceedingly rare hereditary condition produced by a keratin encoding mutation. PC, also known as Jadassohn-Lewandowsky syndrome, is an autosomal dominant genodermatosis distinguished by nail thickening, palmoplantar keratodermas, natal teeth, oral leukokeratosis/leukoplakia, hoarseness, and sebaceous cysts/follicular papules [1-5]. Some authors have suggested that the oral symptoms may appear sooner than lesions of the nails [6]. Bergendal reported that the oral mucosal changes are typical and affect the mucosa, tongue, and the lips [7]. The approximate number of global cases is stated to be between 5,000 and 10,000 [8,9].
Therapy for PC is focused on management of symptoms and improving quality of life. Longevity is not affected, but thickened keratomas on the bottom of the feet can be very painful and incapacitating for everyday ambulation. Recent treatment strategies being studied include statin therapy, rapamycin inhibitors, RNA interference, botulinum neurotoxin, TNF inhibitors, gabapentin, antidepressant SNRIs, and capsaicin [10]. Although most reports imply that oral lesions are common and usually do not need treatment, Gonul et al described a case of intraoral mucosal squamous cell carcinoma in a PC patient with oral leukokeratosis [11]. This mandates the importance of long-term follow-up for all PC patients, asymptomatic or not.
Case Report
A 78-year-old male patient initially presented to the Dental College of Georgia at Augusta University for periodontal implant therapy and prophylaxis appointments. During clinical examination, several incidental clinical findings such as xerostomia, coated tongue, abnormal bilateral white lesions, and hyper-keratinization on the lateral borders of the tongue (Figure 1) and buccal mucosa (Figure 2) were noted. These findings were consistent with patient’s known history of Pachyonychia Congenita, previously affecting his skin, nails (Figure 3), and mucous membranes.
Given the patient’s history and concerning clinicals findings, a consultation was made with the Oral Pathologist and an excisional biopsy on the lateral borders of the tongue and buccal mucosa was recommended to rule out pre-cancerous lesions or other pathological conditions that may be related to the patient’s symptoms.
A punch biopsy of 4 mm diameter and 4mm depth was performed on the patient and the samples were taken from two locations: lateral border of the tongue on the right side and left buccal mucosa. 4-0 chromic gut sutures were used for wound closure and the obtained samples were placed in formalin and sent for pathological evaluation.
Pathological Report
The formalin-fixed tissue specimen was subjected to routine processing after which 5μm sections were cut and stained with hematoxylin and eosin. The tissue sections showed hyperkeratinized surface mucosal epithelium supported by lamina propria (Figure 4). The epithelium was covered by a thick layer of parakeratin. The spinous cell layer was thickened and showed superficial layers of cells exhibiting prominent deeply chromatic nuclei surrounded by vacuolated cytoplasm imparting optically clear cells (Figure 5). The basal and suprabasal layers of cells exhibited the normal cellular maturation pattern without any cellular or nuclear atypia (Figure 6). Subepithelial inflammatory cell infiltrates were noted in the superficial lamina propria. A diagnosis of “Hyperparakeratosis with prominent perinuclear vacuolization of the epithelium”, consistent with Pachyonychia congenita was made.
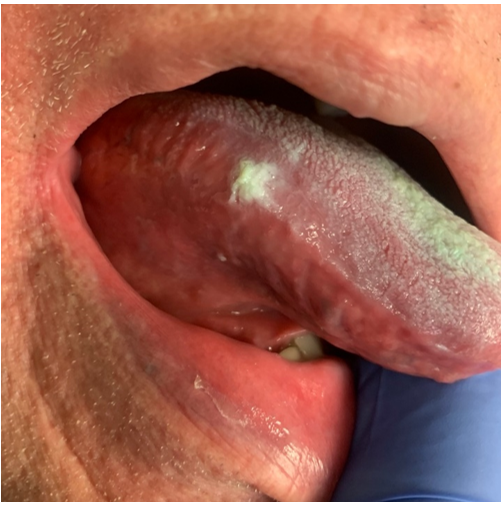
Figure 1: Hyperkeratosis on the lateral border of the tongue
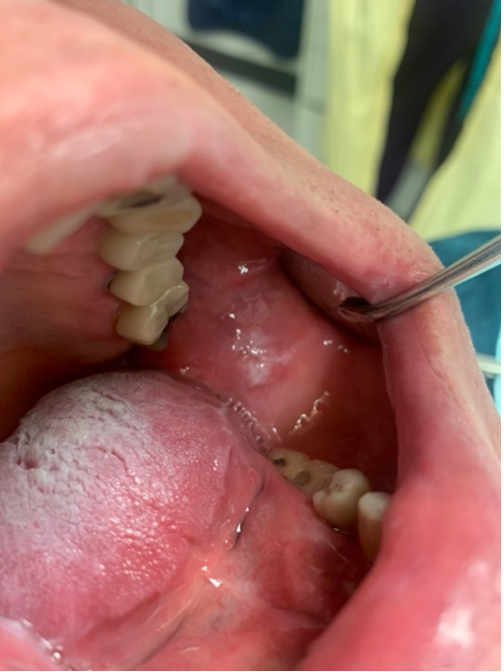
Figure 2: Hyperkeratosis on the buccal mucosa
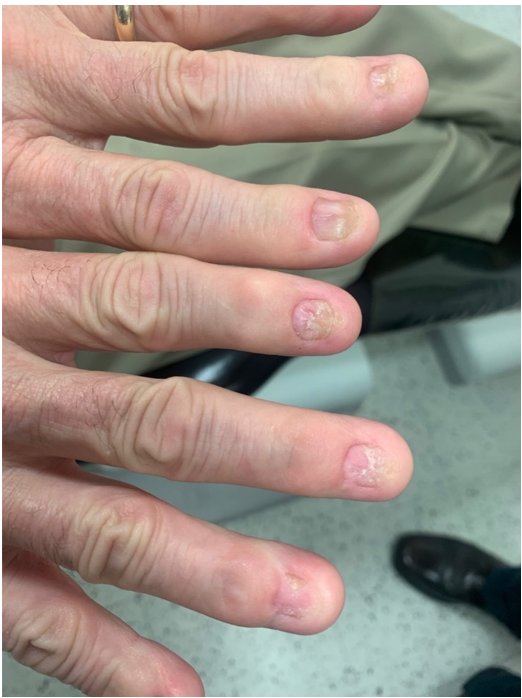
Figure 3: Patient’s fingers showing dystrophic nail beds after losing thickened nail plates
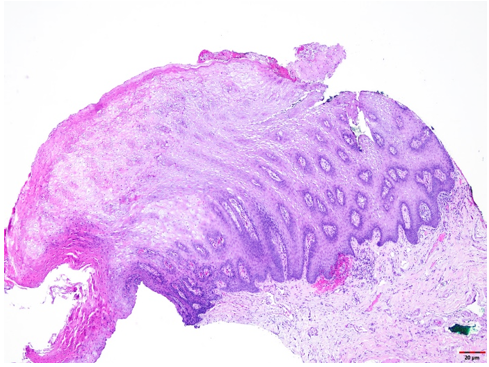
Figure 4: Tissue section exhibiting hyperkeratinized surface mucosal epithelium supported by lamina propria
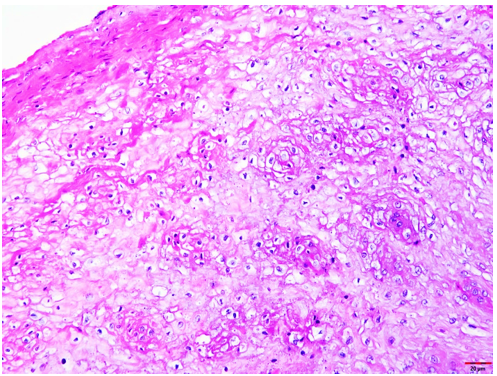
Figure 5: Tissue section exhibiting the thickening of the spinous cell layer with superficial layers of cells exhibiting prominent deeply chromatic nuclei surrounded by vacuolated cytoplasm imparting optically clear cells
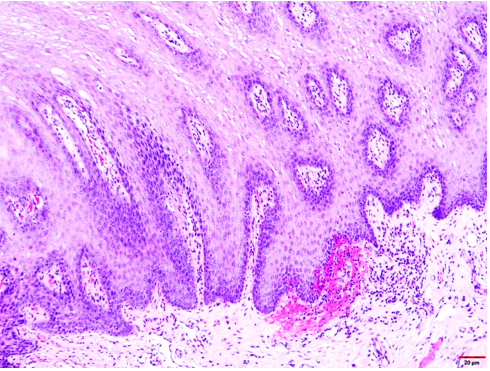
Figure 6: Tissues section exhibiting normal cellular pattern on the basal and suprabasal layers without any cellular or nuclear atypia
Discussion
This report presents a case of Pachyonychia congenita (PC), also known as 'Jadassohn-Lewandowsky syndrome,' a very rare genodermatotic condition characterized by hyper-keratinization disorders. PC exhibits an autosomal dominant mode of inheritance with equal gender distribution and is associated with mutations in genes encoding Keratin 6a/b, 16, or 17 [12]. This condition primarily manifests as massive hyperkeratosis of the distal nail beds, palmoplantar keratoderma, hyperhidrosis, and various cutaneous and mucosal lesions. In this case, we have documented a patient with intra-oral and extra-oral manifestations of PC, highlighting the importance of early identification and management to improve the patient's quality of life.
PC follows an autosomal dominant mode of inheritance, with a high incidence in specific geographic regions and populations, reportedly Croatia and Slovenia with a predominance in Jewish populations [13]. Patients with PC often present with a range of clinical manifestations, which may include, hyperkeratosis of the distal nail beds of both fingers and toes leading to nail plate elevation and apparent thickening, palmoplantar hyperkeratosis, hyperhidrosis, follicular keratosis, xerosis (dry skin), and verrucous lesions on elbows, knees, or lower legs. PC can also affect other areas of the body, including the eyes, ears, nose, and larynx, potentially leading to partial blindness, deafness, and hoarseness. Oral findings may include leukokeratosis oris, characterized by generalized rough white plaques in the oral cavity [1-5].
Histological examination was conducted to rule out comorbidities and confirm leukokeratosis, as in contrast to dyskeratosis congenita there is a consensus that malignant transformation in PC is not common [14]. While there is no cure or specific treatment for PC, histopathological evaluation is essential to confirm the diagnosis and rule out other potential conditions. Recent studies revealed that hyperactivity of EGFR (epidermal growth factor receptor) signaling may lead to processes that induce painful lesions in PC [9,15], and oral erlotinib, a kinase inhibitor that blocks human type 1 EGFR, showed a strong reduction in neuropathic pain in three patients treated for 6 to 8 months [15].
PC does not typically affect life expectancy; however, it can significantly impact the patient's quality of life. Clinical symptoms such as pain, reduced mobility, difficulty in sports participation, and prolonged standing, along with psychological effects due to physical limitations, are commonly reported by patients with PC.
Conclusion
In conclusion, Pachyonychia congenita is an exceptionally rare genetic condition with distinctive clinical and pathological features. It can lead to both intra-oral and extra-oral manifestations, necessitating early identification and management to improve the patient's overall quality of life. Although there is no cure for PC, comprehensive care and support can help alleviate some of the associated symptoms and challenges faced by affected individuals. Further research into potential treatments and genetic counseling for affected families remains crucial in managing this condition.
Data Availability Statement: The data that support the findings of this study are available on request from the corresponding author.
Acknowledgments: The authors report no conflict of interest.
Consent: Informed verbal consent was provided by the patient’s parent (guardian/caregiver).
References
- Gupta SK., Deepika., Monika., Singh K. Jadassohn-Lewandowsky syndrome: A rare genodermatosis. J Pakistan Ass Dermatologists. 2016;26(4):389‒391. [Ref.]
- Pinna R., Cocco F., Campus G., Conti G., Milia E., et al. Genetic and developmental disorders of the oral mucosa: Epidemiology; molecular mechanisms; diagnostic criteria; management. Periodontol 2000. 2019;80:12‒27. [PubMed.]
- Hersh SP. Pachyonychia congenita. Manifestations for the otolaryngologist. Arch Otolaryngol Head Neck Surg. 1990;116(6):732‒734. [PubMed.]
- Maser ED. Oral manifestations of pachyonychia congenita. Report of a case. Oral Surg Oral Med Oral Pathol. 1977;43(3):373‒378. [PubMed.]
- Vucicevic‒Boras V., Kotrulia L., Cekic‒Arambasin A., Pirkic A., Vucic M. Pachyonychia congenita. Case report. Minerva Stomatol. 2005;54(11‒12):691‒694. [PubMed.]
- Anneroth G., Isacsson G., Lagerholm B., Lindvall AM., Thyresson N. Pachyonychia congenita. A clinical, histological and microradiographic study with special reference to oral manifestations. Acta Derm Venereol. 1975;55(5):387‒394. [PubMed] [PubMed.]
- Bergendal B. Orodental manifestations in ectodermal dysplasia - A Review. Am J Med Genet Part A. 2014;164A:2465‒2471. [PubMed.]
- Samuelov L., Smith FJD., Hansen CD., Sprecher E. Revisiting pachyonychia congenita: a case‒cohort study of 815 patients. Br J Dermatol. 2020;182:738‒746. [PubMed.]
- Coulombe PA., Orosco A. Inhibiting EGFR signaling holds promise for treating palmoplantar keratodermas. J Invest Dermatol. 2023;143:185‒188. [PubMed.]
- Wu TT., Eldirany SA., Bunick CG., Teng J. Genotype-structurotype-phenotype correlations in pachyonychia congenita patients. J Invest Dermatol. 2021;141(12):2876‒2884. [PubMed.]
- Gonul M., Gul U., Kilic A., Soylu S., Kocak O., et al. A case of pachyonychia congenita with unusual manifestations: an unusual type or a new syndrome? Int J Dermatol. 2015;54:334‒337. [PubMed.]
- Leachman., Sancy A., et al. Clinical and pathological features of pachyonychia congenita. Journal of Investigative Dermatology Symposium Proceedings. 2005;10(1). [PubMed.]
- Kansky A., et al. Pachyonychia congenita (Jadassohn‒Lewandowsky syndrome)-evaluation of symptoms in 36 patients. Archives of dermatological research. 1993;285:36‒37. [PubMed.]
- Neville B., Damm DD., Chi AC., Allen CM. Oral and maxillofacial pathology 4th edition. St. Louis, MO, Westline Industrial Dr: Elsevier Health Sciences. 2015;693‒695. [Ref.]
- Basset J., Marchal L., Hovnanian A. EGFR signaling is overactive in pachyonychia congenita: effective treatment with oral erlotinib. J Invest Dermatol. 2023;143:293‒304. [PubMed.]