>Corresponding Author : Benhaddouga Khadija
>Article Type : Case Report
>Volume : 5 | Issue : 7
>Received Date : 06 July, 2025
>Accepted Date : 16 July, 2025
>Published Date : 20 July, 2025
>DOI : https://doi.org/10.54289/JCRMH2500134
>Citation : Benhaddouga K, Moustatir M, Bouchane H, Chyate FZ, Hayat M, et al. (2025) Cystic Hygroma: 1 Case Report and Review of the Literature. J Case Rep Med Hist 5(7): doi https://doi.org/10.54289/JCRMH2500134
>Copyright : © 2025 Benhaddouga K, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Case Report | Open Access | Full Text
1Resident Physician, Department of Gynecology and Obstetrics, at Ibno Rochd University Hospital, Casablanca, Morocco
2Professor in the Department of Gynecology and Obstetrics at the Ibno Rochd University Hospital in Casablanca, Morocco
*Corresponding author: Benhaddouga Khadija, Resident Physician, Department of Gynecology and Obstetrics, at Ibno Rochd University Hospital, Casablanca, Morocco
Abstract
Cystic hygroma, or cystic lymphangioma, is a rare congenital malformation of the lymphatic system, most often presenting as cystic masses in the cervico-facial region. Although most cases occur in children, adults are also affected. Diagnosis relies on imaging, particularly ultrasound and MRI, to assess the extent and characteristics of the lesion.
Treatment varies from case to case. Surgical excision remains the reference treatment, but it can be delicate and prone to recurrence. Alternatively, less invasive treatments such as sclerotherapy (OK-432, bleomycin, doxycycline) have shown notable efficacy, especially in macrocystic forms.
Keywords: Cystic hygroma, Cystic lymphangioma, Lymphatic malformation, Congenital tumor, Sclerotherapy
Abbreviations:SA: Semaines d'Aménorrhée, Hb: Hemoglobin, Lc: Leukocyte count, Pq: Platelet count, TP: Prothrombin time, TCA: Activated Partial Thromboplastin Time, Fg: Fibrinogen, ASAT: Aspartate Aminotransferase, ALAT: Alanine Aminotransferase, LDH: Lactate Dehydrogenase, CRP: C-Reactive Protein, MRI: Magnetic Resonance Imaging
Introduction
Cystic hygroma, also known as cystic lymphangioma, is a rare congenital malformation of the lymphatic system, characterized by multilocular cystic masses, often located in the cervico-facial region. First described by Redenbacher in 1828, it occurs mainly in children, although cases in adults have been reported [1]. The pathogenesis remains imperfectly elucidated but generally involves an anomaly in the embryonic development of lymphatic vessels. Diagnosis relies on imaging, in particular ultrasound and MRI, while therapeutic management remains a matter of debate, oscillating between surgical excision and sclerotherapy.
Case Report
This is an 18-year-old patient, with no particular pathological history, ptimigravida primiparous, presenting for fetal death in utero on a dysmorphic syndrome type cystic hygroma on a 23SA+1d pregnancy.
On admission: patient conscious 15/15, hemodynamically and respiratorily stable, urine dipstick negative, active fetal movements not perceived.
Obstetrical ultrasound: non-progressive mono-fetal pregnancy, absent cardiac activity, cephalic presentation, fundal placenta. Estimated fetal weight: 603g +/-134g / Dysmorphic syndrome with major malformatons in the form of generalized edema with ascites and pleurisy, cardiac malformation and enormous cystic hygroma (figure 1).
Biological check-up:
Hb :11,2 Lc :8650 pq :208000 TP :136% TCA :21,5 Fg :3,62
Urea : 0,7 creatinine : 5,8 ASAT/ALAT :34/29 LDH :205 CRP :5,06
Patient triggered by propes giving birth to a stillborn of undetermined sex weighing 720g (figure 2)
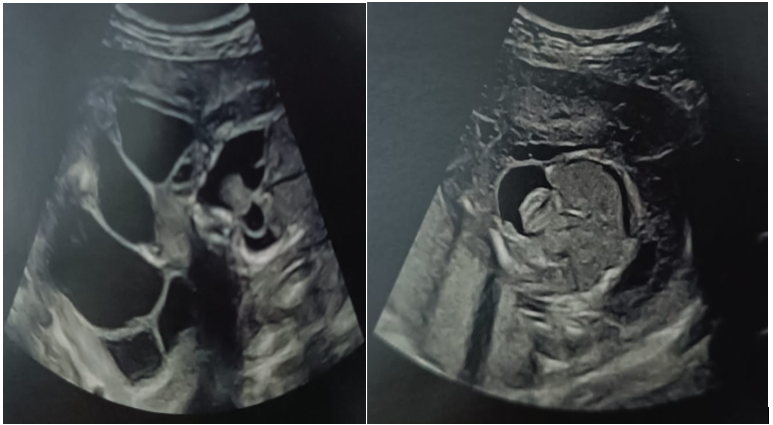
Figure 1: Ultrasound image of dysmorphic syndrome

Figure 2: Image of the polymalformed stillborn
Discussion
Cystic hygroma accounts for around 5-6% of congenital tumours in children, with a predilection for the posterior cervical region [2]. The development of the lesion is often observed from birth or during infancy. However, in some cases, the lesion may remain asymptomatic for years before manifesting itself as a painless swelling, as reported in some adults [3]. Embryologically, cystic hygroma is thought to result from the failure of the connection between the primitive lymph sacs and the venous system, leading to the accumulation of lymph in separate cavities [4].
Diagnosis is essentially based on imaging. Ultrasound can identify the cystic nature of the lesion and assess its relationship with adjacent structures. MRI, however, is the examination of choice for precise mapping of mass extension, particularly in deep or mediastinal forms [5]. On the other hand, puncture for diagnostic purposes should be avoided because of the risk of infection and dissemination.
The treatment of cystic hygroma is the subject of much controversy. Complete surgical excision is often advocated but can prove difficult due to the extension of the lesion and its adhesion to vital structures, notably the nerves and vessels of the neck [6].
Moreover, surgery is associated with a risk of recurrence, especially in cases of incomplete excision. Other less invasive approaches, such as sclerotherapy with OK-432 (picibanil), doxycycline or bleomycin, have shown promising efficacy, particularly for macrocystic lesions [7]. Sclerotherapy has the advantage of minimizing complications and avoiding unsightly scars, which is particularly relevant in children [8].
On the other hand, some mixed or microcystic forms respond less well to this treatment and may require combined management. It should also be noted that, in rare cases, complications such as infection, haemorrhage or airway compression may occur, warranting emergency intervention [9].
In prognostic terms, cystic hygromas generally have a benign course, but recurrence is not uncommon, particularly after partial treatment or in deep-seated forms. Long-term follow-up is therefore recommended, with regular clinical and radiological checks [10].
Conclusion
Cystic hygroma is a benign but potentially complex lymphatic malformation, both diagnostically and therapeutically. Knowledge of its clinical, radiological and evolutionary particularities is essential to adapt its management. While surgery remains the treatment of choice, advances in sclerotherapy now offer a less invasive and often effective alternative. A multidisciplinary approach and rigorous follow-up help optimize prognosis and reduce the risk of recurrence.
Reference
- Redenbacher G. Initial report on lymphatic cysts in the neck. J Anat Pathol. 1828. [Ref.]
- Smith RJ, Burke DK. Cystic hygroma: current surgical management. Laryngoscope. 1991; 101(2):219–24. [Ref.]
- Kennedy TL. Cystic hygroma-lymphangioma: a rare and still unclear entity in adults. Otolaryngol Clin North Am. 1995; 28(5):905–19. [PubMed.]
- Wiegand S, Eivazi B, Barth PJ, et al. Pathogenesis of lymphatic malformations. Otolaryngol Head Neck Surg. 2008; 138(6):772–7. [Ref.]
- Elie C, Garel C, Scheinmann P, et al. Role of MRI in the evaluation of cervicofacial lymphangiomas. Eur Radiol. 2000; 10(4):672–8. [Ref.]
- Fliegelman LJ, Friedland D, Brandwein M, Rothschild MA. Recurrent cystic hygroma: surgical strategies and outcomes. Arch Otolaryngol Head Neck Surg. 1998;124(1):88–92. [Ref.]
- Ogita S, Tsuto T, Nakamura K, Deguchi E, Tokiwa K, Takahashi T. OK-432 therapy for lymphangiomas in children. J Pediatr Surg. 1994; 29(6):784–6. [PubMed.]
- Sainsbury DC, Kessell G, Fall AJ, et al. Long-term results of bleomycin injection therapy for lymphatic malformations. Br J Plast Surg. 2007; 60(4):409–13. [Ref.]
- Bisdorff A, Mulliken JB, Fishman SJ, Burrows PE. Lymphatic malformations: risk of hemorrhage and infection. Ann Otol Rhinol Laryngol. 2005; 114(7):498–504. [Ref.]
- Smith MC, Zimmerman MB. Long-term outcome of lymphatic malformations: a follow-up study. Pediatrics. 2004; 114(3):e494–8. [Ref.]