>Corresponding Author : Benhadouga Khadija
>Article Type : Case Report
>Volume : 5 | Issue : 6
>Received Date : 02 July, 2025
>Accepted Date : 12 July, 2025
>Published Date : 16 July, 2025
>DOI : https://doi.org/10.54289/JCRMH2500129
>Citation : Benhaddouga K, Fatima ET, Bahlioui FZ, Benchrifi Y, Benhassou M, et al. (2025) Ovarian Fibrosarcoma: 1 Case Report and Review of the Literature. J Case Rep Med Hist 5(6): doi https://doi.org/10.54289/JCRMH2500129
>Copyright : © 2025 Benhadouga K, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Case Report | Open Access | Full Text
1Resident Physician, Department of Gynecology and Obstetrics, at Ibn Rochd University Hospital, Casablanca, Morocco
2Professor in the Department of Gynecology and Obstetrics at the Ibno Rochd University Hospital in Casablanca, Morocco
*Corresponding author: Benhadouga Khadija, Resident Physician, Department of Gynecology and Obstetrics, at Ibno Rochd University Hospital, Casablanca, Morocco
Abstract
Ovarian fibrosarcoma is an extremely rare malignant tumor, accounting for less than 1% of all ovarian tumors. It is characterized by a proliferation of spindle-shaped cells with high mitotic activity, often posing a diagnostic challenge due to its resemblance to certain benign tumors. Diagnosis is based on histopathological analysis, possibly supplemented by immunohistochemistry. Treatment is essentially surgical, while the role of chemotherapy remains controversial in the absence of consensus. Prognosis depends on histological aggressiveness and stage at diagnosis. This paper summarizes current knowledge of this rare entity and highlights the need for better data collection through multicenter studies.
Keywords: Ovarian Fibrosarcoma; Mesenchymal Tumor; Sarcoma; Rare Tumor; Ovary; Histology; Surgical Treatment
Introduction
Ovarian fibrosarcoma is an extremely rare malignancy, accounting for less than 1% of malignant neoplasms of the ovary. It is a primitive mesenchymal tumor derived from ovarian connective tissue. It occurs mainly in young to middle-aged women, often between the ages of 20 and 50 [1]. Clinically, symptoms are unspecific, complicating the initial diagnosis. A pelvic mass, abdominal pain or enlargement of the abdomen may be reported. Pelvic imaging usually reveals a solid, sometimes heterogeneous ovarian mass. However, only histopathological examination can confirm the diagnosis.
Case Report
This is a 35-year-old female patient, nulligravida, with no particular pathological history, presenting with chronic pelvic pain with abdominal distension.
Her history of illness goes back 8 months, with chronic pelvic pain of the heaviness type with abdominal distension progressively increasing in volume, evolving for a month. All this in a context of deteriorating general condition.
On clinical examination: Abdominal distension with diffuse dullness, positive float and icicle signs.
Abdominal ultrasound: Presence of two solidocystic latero-uterine masses measuring 11x7 cm on the right and 18x9 cm on the left, associated with a peritoneal effusion at the level of the douglas and multiple peritoneal masses, as well as a bilateral pleural effusion of moderate abundance, predominantly on the right (figure1).
On thoracoabdomino-pelvic CT:
- Ascites of moderate abundance peri-hepatic and peri-splenic, parieto-colic gutter, pelvis and inter-annulus.
- Infiltrated appearance of abdominopelvic fat in flames, predominantly anterior, supra- and submesocolic.
- Absence of peritoneal nodule
- Large solid cystic abdominopelvic mass above and laterally in the right uterus measuring 20.6 x13x25.3 cm, most likely of right ovarian origin.
- The left ovary is the site of a regularly contoured oval mass measuring 30x28x32 mm.
- Several subcentimetric iliac lymph node formations with no signs of atypia, the largest measuring 4 mm in minor axis
- Moderate to large bilateral pleural effusion
CA125: 478 UI/mL
The patient underwent bilateral adnexectomy. Anatomopathological study: Morphological and histochemical appearance compatible with ovarian fibrosarcoma.
Discussion
Diagnosis is based on microscopic analysis, which shows a proliferation of spindle-shaped cells arranged in intersecting bundles, with marked mitotic activity, often exceeding 4 mitoses per 10 fields at high magnification [1,2]. It is important to distinguish this entity from other fibroid tumours of the ovary, notably mitotically active fibroma or thecosarcomas, which may present a similar morphological appearance. Immunohistochemistry can sometimes help to exclude other tumor types. In the absence of specific markers, diagnosis is based on strict morphological criteria, notably cellular atypia and mitotic activity [3].
Treatment is mainly based on complete surgical excision. In young women, unilateral salpingo-oophorectomy may be considered if the tumour is localised. On the other hand, in the case of advanced disease or in women who do not wish to become pregnant, more radical surgery including total hysterectomy and bilateral adnexectomy is often performed [4]. The role of chemotherapy or radiotherapy remains uncertain, given the very limited number of cases described in the literature. However, ifosfamide- and doxorubicin-based protocols have been proposed for certain aggressive forms, particularly in cases of recurrence or residual postoperative disease [4,5].
The prognosis of ovarian fibrosarcoma is variable. In well-differentiated forms confined to the ovary, the evolution may be relatively favorable if the tumor is completely resected. In contrast, forms with high mitotic activity, capsular invasion or peritoneal dissemination are associated with a high risk of recurrence and a poor prognosis [2,5]. The absence of standardized recommendations and the small number of cases mean that an individualized approach is required, ideally discussed in a multidisciplinary consultation meeting.
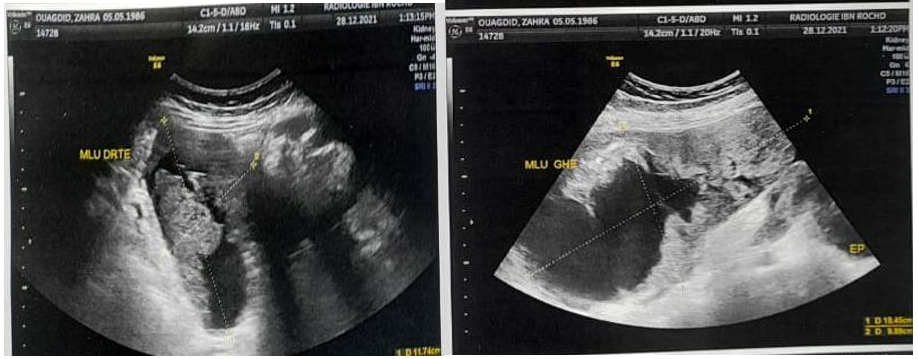
Figure 1: Ultrasound image of lateri-uterine masses
Conclusion
Ovarian fibrosarcoma is a rare but potentially aggressive tumor. Diagnosis is based exclusively on histological analysis, requiring anatomopathological expertise. Treatment is mainly surgical, with adjuvant options yet to be defined. The lack of prospective data underscores the need to publish cases and set up registries to improve knowledge of this pathology.
Reference
- Prat J. Ovarian fibrosarcomas: a clinicopathologic analysis of 15 cases. Cancer. 1982;50(10):2193–2202. [Ref.]
- Young RH. Ovarian fibrosarcoma: a clinicopathologic analysis of 25 cases and review of the literature. Am J Surg Pathol. 1984;8(5):345–358. [Ref.]
- Zaloudek CJ., Norris HJ. Mesenchymal tumors of the ovary. In: Kurman RJ, ed. Blaustein's Pathology of the Female Genital Tract. 4th ed. Springer. 1994. [Ref.]
- Abu-Rustum NR., et al. Ovarian fibrosarcoma: a case report and review of the literature. Gynecol Oncol. 2002;85(1):181–184. [Ref.]
- Yamamoto T., et al. Ovarian fibrosarcoma with rapid progression. J Obstet Gynaecol Res. 2014;40(1):241–245. [Ref.]