>Corresponding Author : Elomri Hajar
>Article Type : Case Report
>Volume : 4 | Issue : 9
>Received Date : 19 June, 2024
>Accepted Date : 01 July, 2024
>Published Date : 16 July, 2024
>DOI : https://doi.org/10.54289/JCRMH2400135
>Citation : Hajar E, Errih L, Imane Z, Fadwa B, Mohamed J, et al. (2024) A Rare Case of an Autoimmune Polyglandular Syndrome Type 2 During Pregnancy. J Case Rep Med Hist 4(9): doi https://doi.org/10.54289/JCRMH2400135
>Copyright : © 2024 Hajar E, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Case Report | Open Access | Full Text
1Resident Physician, Department of Gynecology and Obstetrics, at Ibn Rochd University Hospital, Casablanca, Morocco
2Professor in the Department of Gynecology and Obstetrics at the Ibn Rochd University Hospital in Casablanca, Morocco
*Corresponding author: Elomri Hajar, Resident Physician, Department of Gynecology and Obstetrics, Ibno Rochd University Hospital, Casablanca, Morocco
Abstract
Autoimmune polyglandular syndromes (APS) are a rare heterogeneous disorders characterized by combining two or more organ-specific endocrinopathies. During pregnancy, only a few cases of these syndromes have been described. We report a case of autoimmune polyglandular syndrome type 2 presenting during pregnancy and complicated in the immediate postpartum by a severe polyserositis.
Keywords: Autoimmune, Polyglandular, Endocrinopathies, Postpartum.
Abbreviations: APS: Autoimmune polyglandular syndromes
Introduction
Autoimmune polyglandular syndromes (APS) are rare inherited disorders characterized by multiple endocrine dysfunction. Based on clinical presentation, they are classified into three types. Types 1 and 2 are well-characterized entities, whereas type 3 is poorly characterized [1]. The presence of lymphocytic infiltration of the affected glands, organ-specific autoantibodies in serum, and an association with HLA linkage genes support the autoimmune nature of these disorders [1]. As these patients have more than one endocrine dysfunction, it is important to look for this syndromic grouping, particularly during pregnancy, in the case of any parturient presenting with a group of major endocrinopathies, because of the predominant maternal-fetal risk, and in anticipation of comprehensive management. We report a case of a 31 years old patient with an autoimmune polyglandular syndrome type 2 presenting during pregnancy and complicated in the immediate postpartum by a severe polyserositis.
Case Report
Mrs K.M , 31 years old pregnant patient, nulliparous nulligest, was referred by her endocrinologist for a pregnancy monitoring at 12 weeks of amenorrhea with a severe polyglandular pathological history, a type 1 diabetes treated with insulin discovered at the age of 16 years old after a Ketoacidosis crise , hashimoto's thyroiditis on levothyroxin 200 µg per day for 10 years, she is also followed up for addison's disease on cortisolic supplementation for 5 years, with two previous hospitalizations in the endocrinology department for an acute surrenal insufficiency. She also has celiac disease, diagnosed at an early age. There is no family history of autoimmune disease. The pregnancy follow-up was unremarkable during the first and second trimesters , a monthly hormonal check-up was carried out with no abnormalities. A morphological ultrasound was carried out at 23 weeks of amenorrhea , showing a fetus with normal overall morphology and no cardiac renal or skeletal malformations. The evolution of the pregnancy was marked by the onset of glycemic imbalance at 34 weeks of amenorrhea , complicated by fetal macrosomia above the 97th percentile and a major hydramnios , Therefore an hospitalization in the high-risk pregnancy unit was indicated for fetal and maternel monitoring and surveillance and pulmonary maturation with corticoids Injection. The patient underwent an emergency cesarean section for a non-reassuring fetal condition at 35 weeks gestation with abundant ascites on surgical exploration, giving birth to a female , Apgar 8/10, birth weight 4200g with a severe respiratory distress secondary to an hyaline membrane disease , hence the indication for an urgent admission to neonatal intensive care unit for imminent care .
The immediate postpartum period was marked by the onset of a profound hypoglycemia at 0.17 , associated to a cardiorespiratory decompensation , with a consisting diffuse abdominal pain. Physical examination revealed an altered general condition with no fever, blood pressure of 80/60 mm Hg, pulse of 112 beats per minute and respiratory rate of 26 cycles per minute. Abdominal examination revealed significant meteorism with tympany and diffuse abdominal tenderness. On rectal examination, the rectal ampulla was empty. The gynaecological examination revealed a clean caesarean section scar, minimal lochia, and a uterus that was difficult to assess due to abdominal distension. The thoracic Abdominopelvic CT scan with injection of contrast medium showed the presence of a polyseritis consisting of a bilateral pleural effusion of low abundance and a pericardial effusion of medium abundance, and ascites of high abundance which was punctured twice.
The biological results showed a low hypoalbuminemia at 18 g/l treated with venous albumin infusion. The patient was discharged at 17 days postpartum, after a 10-day stay in the obstetric reanimation unit. As for the newborn, he was discharged after 8 days in the neonatal intensive care unit. The care management was relayed to the endocriology department for a strict readaptation of the hormonal treatment.
Discussion
Autoimmune polyglandural syndrome (APS), can affect several endocrine glands in the same patient, hence the term “polyglandular dysfunction”. They are classified into three types (Figure 1).
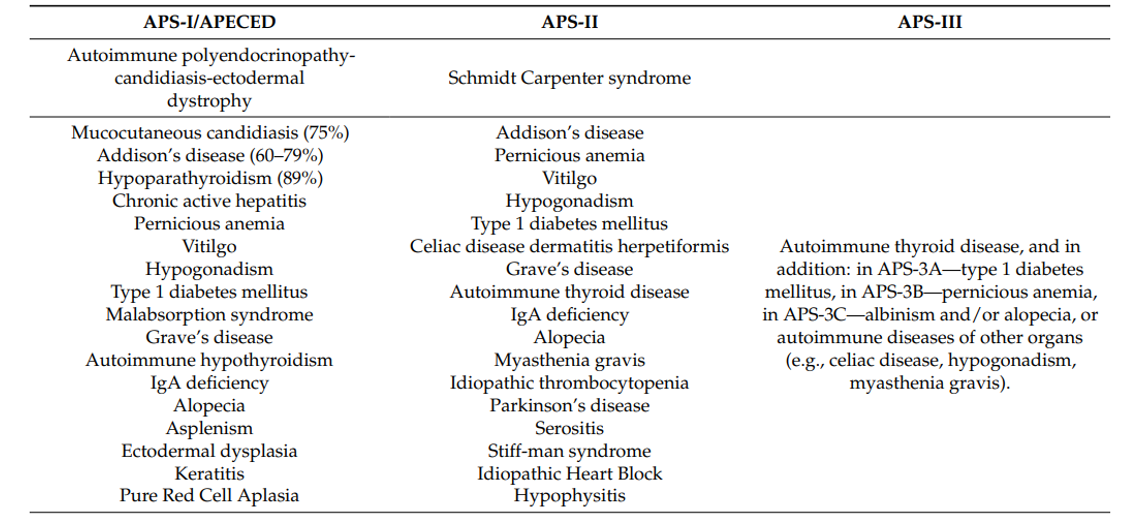
Figure 1. Autoimmune polyendocrine syndromes [2]
These disorders are generally characterized by the presence of circulating organ-specific antibodies, even in the absence of overt clinical disease [1]. The term “polyglandular autoimmune syndrome” was first introduced by Neufeld , and classification into three subtypes is based on specific clinical features [3].
Type I autoimmune polyendocrinopathy consists of a triad associating Addison's disease, chronic mucocutaneous candidiasis and hypoparathyroidism, several disorders may be associated, and the onset of symptoms occurs at an early age, usually during the first decade of life, with males and females equally affected [4]. In type I, there appears to be no correlation with HLA, although familial clustering of cases does exist, occurring through autosomal recessive inheritance [5].
Polyendocrinopathy type II is the most common form of polyendocrine dysfunction [6]. APS type 2 is still a relatively rare condition, with an estimated prevalence of five per 100,000 in the United States [1]. The type II classification, or Schmidts syndrome, requires the presence of Addison's disease, autoimmune thyroiditis and/or insulin-dependent diabetes mellitus. The disorder may also be associated with biermer anemia, gonadal insufficiency or celiac disease [7]. Unlike APS type I, APS type II has a later age of presentation, with a maximum age of 30 years. There is a pronounced female predominance, with a dominant mode of inheritance involving the important genes found with the HLA complex on chromosome 6, in particular the HLA-DR genes DR3 and DR4 [8].
Autoimmune polyglandular syndrome type 3 is characterized by the occurrence of autoimmune thyroiditis accompanied by other organ (non-ovarian specific) autoimmune disorders in the absence of adrenal insufficiency [2].
It can be difficult to recognize the manifestations of APS 2 in a pregnant woman because there is a significant overlap between the physiologic changes associated with pregnancy and the non-specific symptoms of APS 2 , on the other hand, pregnancy is a physiologically immune-tolerant state [9]. Very few cases of APS 2 have been described during pregnancy. In the first trimester, APS 2 can complicate pregnancy and be confused with hyperemesis gravidarum as a cause of hypoglycemia and electrolyte imbalance [10].
Biologically, a number of organ-specific autoantibodies are present in serum, including antisurenal, antithyroid and antipancreatic antibodies [11].
The treatment of APS is individualized and based on a correction of the main abnormalities. In patients with hypothyroidism and Addison's disease, thyroid supplementation therapy can precipitate a life-threatening addisonian crisis ,it is therefore vital to restore the surrenal function in hypothyroid patients if an autoimmune polyendocrine syndrome is suspected before starting replacement therapy. Thyroid function often improves once glucocorticoid replacement is started [12].
The prognosis of autoimmune polyendocrinopathies will depend on the impact of all endocrinopathies combined on the course of pregnancy; therefore, early diagnosis of the type of autoimmune polyendocrinopathy, particularly during pregnancy, is important, not only to initiate adequate replacement therapy for the organs thus affected, but also to carry out genetic analyses [13]. In families where the syndrome has been documented, relatives should be notified of the first signs and symptoms. In this at-risk population, as well as in patients with multiple disorders, screening every 3 to 5 years should take place with blood glucose measurements, thyroid function tests, B12 levels and cortisol levels, as such screening can prevent significant morbidity and mortality in these patients and their descendants.
Conclusion
Autoimmune polyglandular syndromes are rare disorders characterized by the development of multiple endocrine dysfunction of target organs. The syndromes encompass a group of diseases characterized by poly-glandular dysfunction. These syndromes should be considered when dealing with pregnant patients with multi-glandular endocrine dysfunction, because the maternal and fetal , vital and functional prognosis is at risk.
References
- Majeroni BA, Patel P. Autoimmune polyglandular syndrome, type II. Am Fam Physician. 2007;75:667-70. [PubMed.]
- Komorowska B. Autoimmune premature ovarian failure. Menopausal Rev. 2016;4:210-4. [PubMed.]
- Neufeld M, Maclaren N, Blizzard R. Autoimmune polyglandular syndrome. Pediatr Ann. 1980;9:154-62. [PubMed.]
- Neufeld M, Maclaren N, Blizzard R. Two types of autoimmune Addison's disease associated with different polyglandular syndromes. Medicine (Baltimore). 1981;60:355-62. [PubMed.]
- Khalil S, Evers M. Case report: Autoimmune polyglandular syndrome. N J Med. 1995;92:10. [PubMed.]
- Riley WJ. Autoimmune polyglandular syndromes. Horm Res. 1992;38(Suppl 2):9-15. [PubMed.]
- Graber MA, Freed HA. Polyglandular autoimmune syndrome: A cause of multiple and sequential endocrine emergencies. Am J Emerg Med. 1992;10:130-2. [PubMed.]
- Maclaren NK, Riley WJ. Inherited susceptibility to autoimmune Addison's disease is linked to human leukocyte antigens DR3 and/or DR4 except when associated with type 1 autoimmune polyglandular syndrome. J Clin Endocrinol Metab. 1986;62:455-9. [PubMed.]
- Kaaja RJ, Greer IA. Manifestations of chronic disease during pregnancy. JAMA. 2005;294:2751. [Ref.]
- Krysiak R, Samborek M. Coexistence of autoimmune polyglandular syndrome type 2 and diabetes insipidus in pregnancy. Am J Med Sci. 2011;342:433-4. [PubMed.]
- Caterio AZ, Wagner R, Gill JS. Organ specific cardiac antibodies: Serological markers for systemic hypertension in autoimmune polyendocrinopathy. Lancet. 1991;337:1111-5. [PubMed.]
- Liu MM, Reidy AB, Saatee S, Collard CD. Perioperative steroid management: approaches based on current evidence. Anesthesiology. 2017;127:166. [PubMed.]
- Husebye ES, Anderson MS, Kämpe O. Autoimmune polyendocrine syndromes. N Engl J Med. 2018;378:1132-41. [PubMed.]