>Corresponding Author : Ziad Imane
>Article Type : Case Report
>Volume : 4 | Issue : 7
>Received Date : 09 May, 2024
>Accepted Date : 20 May, 2024
>Published Date : 28 May, 2024
>DOI : https://doi.org/10.54289/JCRMH2400134
>Citation : Doha EK, Imane Z, Sarah T, Mohamed J, Amine L, et al. (2024) Prune Belly Syndrome: About Two Cases. J Case Rep Med Hist 4(7): doi https://doi.org/10.54289/JCRMH2400134
>Copyright : © 2024 Doha EK, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Case Report | Open Access | Full Text
1Resident Physician, Department of Gynecology and Obstetrics, at Ibno Rochd University Hospital, Casablanca, Morocco
2Professor in the Department of Gynecology and Obstetrics at the Ibno Rochd University Hospital in Casablanca,Morocco
*Corresponding author: Ziad Imane, Resident Physician, Department of Gynecology and Obstetrics, at Ibno Rochd University Hospital, Casablanca, Morocco
Abstract
Prune belly syndrome is a rare predominantly male congenital disorder, typically associating aplasia or large hypoplasia of the muscles of the anterior wall of the abdomen, urinary malformations and bilateral cryptorchidism. The etiology is not known. It is associated with other congenital anomalies and whose evolution varies from stillbirth due to major renal and respiratory dysplasia to a practically normal child.
Keywords: Prune-Belly Syndrome; Cryptorchidism; Urinary Tract Dilation; Deficient Abdominal Wall
Abbreviations: PBS: Prune Belly Syndrome
Introduction
Prune Belly syndrome or Eagle-Barrett syndrome was first described by the German Frolich in 1839. It was not until 1950 that Osler named it "Prune Belly". It is a predominantly male congenital disorder typically associated with aplasia or large hypoplasia of the muscles of the anterior wall of the abdomen, urinary malformations and bilateral cryptorchidism [1,2]. 75% of cases are associated with pulmonary, osteoarticular, cardiac and gastrointestinal malformations [2]. There are two clinical forms, complete and partial, whose evolution varies from stillbirth due to major renal and respiratory dysplasia to a practically normal child.
We report 2 cases of this rare congenital disorder to create awareness about the early diagnosis and the early management of PBS.
Case Report
Case 1:
Male newborn, from an unattended pregnancy carried to term, caesarean delivery, front presentation, Apgar 7/10. Mother aged 26 years, G2P0, 2 unexplored abortions, diabetic on insulin, negative infectious anamnesis, no notion of consanguinity. Clinical examination revealed a pink, tonic, responsive newborn with a sucking reflex, weight 3300g, height 52cm and head circumference 36cm. Abdominal examination revealed a thinned, flaccid and wrinkled abdominal wall, with palpation of the intestinal loop subcutaneously (Figure 1). Osteoarticular examination revealed clubfoot (Figure 2). Cardiovascular, pleuropulmonary and abdominal examinations were unremarkable. Paraclinical examinations included: a chest X-ray showing thoracic and spinal deformity, with a cardiothoracic index of 0.67. Abdominal and pelvic ultrasound revealed two kidneys of respected size and parenchyma, bilateral pyelocaliceal dilatation, a highly dilated ureter (bilateral mega-ureter), a thickened bladder wall in replenishment with anterior abdominal muscle hypoplasia and bilateral cryptorchidism. Biological tests were unremarkable, with correct renal function.
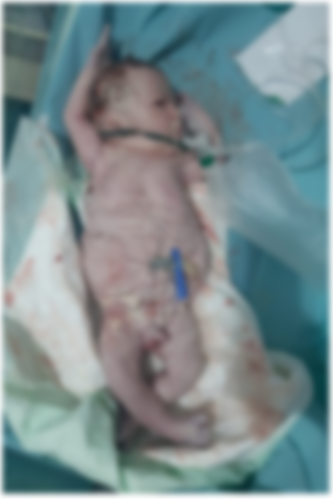
Figure 1. Appearance of the newborn with PBS: wrinkled, redundant skin with bulging at the flanks due to deficient of abdominal wall musculature
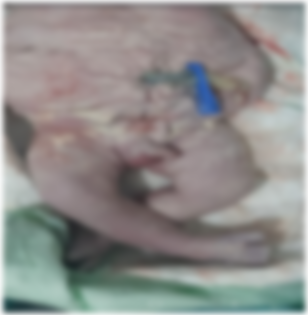
Figure 2. Appearance of the newborn with PBS: Clubfoot
Case 2:
Male newborn, from an unattended pregnancy carried to term, Caesarean delivery, breech presentation, Apgar 6/10. Mother aged 39 years, G3P3, no particular pathological history, negative infectious anamnesis, no notion of consanguinity. Clinical examination revealed a pink, tonic, responsive newborn with a sucking reflex, weight 3570g, height 54cm and head circumference 36cm. Abdominal examination revealed a thinned, flaccid and wrinkled abdominal wall, with palpation of the intestinal loop subcutaneously (Figure 3).
Osteoarticular, cardiovascular and pleuropulmonary examinations were unremarkable. Paraclinical examinations included: a chest X-ray showing thoracic and spinal deformity, with a cardiothoracic index of 0.67. Abdominal and pelvic ultrasound revealed two kidneys of respected size and parenchyma, bilateral pyelocaliceal dilatation, a highly dilated ureter (bilateral mega-ureter), a thickened bladder wall in replenishment with anterior abdominal muscle hypoplasia, and bilateral cryptorchidism with moderate ascites. Biological tests were unremarkable, with correct renal function.

Figure 3. Newborn male with PBS, demonstrating classic abdominal wall appearance
Discussion
Prune Belly Syndrome (PBS) is an extremely rare anatomo-radiological syndrome with an estimated incidence of one case in 40,000 births, and 95% of patients are male [2].
Although the exact mechanism is still unclear, the pathogenesis is mainly linked to urethral obstruction or a defect in mesodermal development. Urethral obstruction early in development causes massive distension of the bladder and urinary ascites, leading to underdevelopment of the abdominal wall musculature and preventing testicular descent. Micturition disorders lead to oligohydramnios, and depending on severity, pulmonary hypoplasia and Potter's sequence and they have a profound and lasting impact on renal, ureteral and bladder function. Most cases are sporadic, a few cases linked to mutations in the CHRM3 gene (at 1q43) have been observed in certain siblings. Several candidate genes have been suggested, but there is insufficient evidence to suggest a monogenic disease. Copy number variations may be the cause of unexplained genetic cases of PBS [3].
Prune belly syndrome (PBS) is often identified via antenatal examination, during routine ultrasonography, and manifests as oligohydramnios and massive bladder dilatation, mild to severe bilateral hydroureteronephrosis, fetal ascites and, occasionally, renal dysplasia and urachus diverticulum. Other anomalies include cryptorchidism, pulmonary hypoplasia, clubfoot and malformations characteristic of the Potter sequence [4]. Other malformations should always be checked pre- and post-natally, namely pulmonary, cardiac, skeletal, gastrointestinal and genital malformations. These malformations have been reported by Routh et al, with an incidence of 25% for cardiovascular, 24% for gastrointestinal, 23% for musculoskeletal, 58% for respiratory and 15% for genital [2].
Postnatal diagnosis is based on abdomino-pelvic ultrasound, supplemented by abdomino-pelvic CT scan, trans-thoracic ultrasound to check for cardiac malformations, renal work-up to assess renal function, hip ultrasound with skeletal X-ray to check for skeletal malformations, and karyotype to check for deletion of nuclear factor 1-beta (HNF1beta) [1,5].
Management begins with ultrasound monitoring of the urinary tract and amniotic fluid volume throughout the pregnancy, in case of diagnostic doubt. Early decompression of the severe bladder outlet obstruction that contributes to oligohydramnios is recommended. Antibiotic prophylaxis should begin at birth, but the mainstay of treatment remains surgery: abdominoplasty, orchidopexy and urinary tract reconstruction [6,7]. For patients with mild abdominal wall dysplasia, postures are acceptable and do not require abdominoplasty. However, for severe cases, surgical treatment is discussed on a case-by-case basis. Pyelostomy, ureterostomy and cystostomy are also undertaken to shunt urine temporarily in certain unstable infants who cannot tolerate the surgical procedure. Renal transplantation is sometimes unavoidable for patients with renal failure.
The postnatal course of infants with PBS depends on renal function and comorbidities, and risk is determined accordingly. The risk of severe renal dysfunction and pulmonary hypoplasia is very high in 20% of patients; in 40% of patients with classic PBS features, the prognosis depends on the degree of renal dysplasia, with 4% at risk of acute renal failure. The 40% of patients at low risk have normal renal function and mild phenotypic features. Prematurity is common, with cardiovascular and pulmonary problems. Estimated perinatal mortality rates in PBS range from 10 to 25% [8,9].
Conclusion
Prune Belly syndrome is characterized by the triad of aplasia or large hypoplasia of the anterior abdominal wall muscles, dilatations of the urinary tract and bilateral cryptorchidism. It is imperative to look for other malformations that may be associated. The prognosis of SBP patients varies according to the severity of pulmonary and urinary tract hypoplasia, which are sometimes fatal.
References
- Xu W, Wu H, Wang D-X, Mu Z-H. (2015) A Case of Prune Belly Syndrome. Pediatr Neonatol. 56(3): 193-196. [Ref.]
- Fette A. (2015) Associated rare anomalies in prune belly syndrome: a case report. Journal of Pediatric Surgery Case reports. Elsevier. 3(2): 65-71. [Ref.]
- Diao B, Diallo Y, Fall PA, Ngom G, Fall B, et al. (2008) Syndrome de Prune Belly?: aspects épidémiologiques, cliniques et thérapeutiques. Prog En Urol. (18): 470-474. [Ref.]
- Grover H, Sethi S, Garg J, Ahluwalia AP. (2017) Pseudo Prune Belly syndrome: diagnosis revealed by imaging? A case report and brief review. Pol J Radiol. 82: 252-257. [PubMed.]
- Granberg CF, Harrison SM, Dajusta D, Zhang S, Hajarnis S, et al. (2012) Genetic basis of prune belly syndrome: screening for HNF1? gene. J Urol. 187(1): 272-278. [PubMed.]
- Seidel NE, Arlen AM, Smith EA, Kirsch AJ. (2015) Clinical manifestations and management of Prune Belly syndrome in a large contemporary pediatric population. Urology. 85(1): 211-215. [PubMed.]
- Dénes FT, Park R, Lopes RI, Moscardi PRM, Srougi M. (2015) Abdominoplasty in Prune Belly Syndrome. J Pediatr Urol. 11(5): 291-292. [PubMed.]
- Tibor-Denes F, Tavares A, Machado M, Giron A, Srougi M. (2017) Gonadal function and reproductive system anatomy in post puberal prune belly syndrome patients. The Journal of Urology. 197(4S). [Ref.]
- Gallo C, Costa W, Favorito L, Sampaio F. (2017) Prune belly syndrome: is penile structures similar to normal fetuses? In: State University of Rio de Janeiro, Urogenital Research Unit, Rio de Janeiro, Brazil. London, United Kingdom. [Ref.]