>Corresponding Author : Zahri S
>Article Type : Case Report
>Volume : 3 | Issue : 10
>Received Date : 11 Dec, 2023
>Accepted Date : 26 Dec, 2023
>Published Date : 30 Dec, 2023
>DOI : https://doi.org/10.54289/JCRMH2300147
>Citation : Zahri S, Belhakim M, Wakil N, Arous A, Haboub M, et al. (2023) Unifying the Puzzle: Cardiac Amyloidosis in the Spectrum of Generalized Edematous Syndromes. J Case Rep Med Hist 3(10): doi https://doi.org/10.54289/JCRMH2300147
>Copyright : © 2023 Zahri S, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Case Report | Open Access | Full Text
1University Hospital Center Ibn Rochd, Hospital Street, 20360 Casablanca, Morocco
2Cardiology department UHC Ibn Rochd, Casablanca
*Corresponding author: Zahri Soukaina and Mehdi Belhakim, University Hospital Center Ibn Rochd, Hospital Street, 20360 Casablanca, Morocco
Abstract
Introduction: Addressing the diagnostic challenges posed by nephrotic syndrome and congestive heart failure, this article underscores the pivotal role of comprehensive investigations in unraveling underlying etiologies. Amid the diverse spectrum of potential causes, the focus is directed towards amyloidosis—a distinct category characterized by abnormal amyloid fibril accumulation in organs. Despite its infrequency, the gravity and potential for targeted interventions accentuate the importance of considering amyloidosis in the differential diagnosis.
Case Presentation: A detailed case study of a 68-year-old female with generalized edematous syndrome revealed a complex clinical scenario involving cardiac and renal manifestations. AL amyloidosis was confirmed through thorough diagnostic assessments, guiding a targeted therapeutic approach.
Discussion: The discussion delves into the pathophysiology of AL amyloidosis, diagnostic considerations, and the significance of ECG and echocardiographic findings. It highlights the importance of laboratory assessments, especially immunologic tests, and emphasizes the role of biopsy in confirming the diagnosis.
Conclusion: The complexity of AL amyloidosis is underscored, emphasizing the interplay of cardiac, renal, and hematologic aspects. Early diagnosis and a tailored multidisciplinary therapeutic approach yielded positive outcomes, contributing valuable insights to the evolving understanding of amyloidosis management.
Keywords: Congestive Heart Failure; Nephrotic Syndrome; Amyloidosis
Abbreviations: ECG: Electrocardiogram, eGFR: Estimated Glomerular Filtration Rate, BNP: B-Type Natriuretic Peptide, ATTR: Transthyretin Amyloidosis, Al Amyloidosis: Amyloid Light Chain or Primary Amyloidosis, GLS: Global Longitudinal Strain
Background
Diagnosing patients solely with nephrotic syndrome or congestive heart failure is insufficient in providing a comprehensive understanding of the underlying etiology. Clinicians are encouraged to pursue thorough investigations to identify the root causes of these syndromes, as this approach significantly informs patient management and prognosis [1]. Among the myriad potential etiologies, amyloidosis represents a distinctive and noteworthy category of diseases characterized by the aberrant accumulation of amyloid fibrils in various organs [2]. Despite its rarity, the gravity and potential for curative interventions underscore the importance of considering amyloidosis in the differential diagnosis [3].
Case Presentation
We present a comprehensive case study of a 68-year-old female patient with no notable medical history, admitted to our department for a generalized edematous syndrome. The syndrome initially manifested in the lower limbs before extending to the face and upper extremities, accompanied by worsening dyspnea initially on exertion, later at rest, within the context of a progressive decline in overall health. The electrocardiogram (ECG) revealed diffuse low voltage with right bundle branch block (Figure 1). Echocardiography demonstrated features consistent with symmetrical hypertrophic cardiomyopathy affecting the septum and posterior wall (Figure2). Despite preserved ejection fraction, global longitudinal strain was impaired, sparing the apex and presenting a characteristic rosette-like appearance (Figure3). Pulmonary hypertension at 60 mmHg was noted (Figure4), impacting the right heart chambers, along with a moderate pericardial effusion. Biological assessments unveiled an impure nephrotic syndrome, comprising 4 g/24h proteinuria, hypoalbuminemia (25 g/L), hypoproteinemia (54 g/L), hematuria, and renal insufficiency with an estimated glomerular filtration rate (eGFR) of 35 ml/min. Hematological analysis indicated normochromic normocytic regenerative anemia. Plasma protein electrophoresis showed no abnormalities, but serum protein immunofixation suggested a monoclonal gammopathy with IgG, along with elevated serum free lambda light chains. We also observed an elevation in the levels of B-type natriuretic peptide (BNP) and troponins. A bone scintigraphy confirmed a Perugini Grade 1 classification. Salivary gland biopsy with immunohistochemistry confirmed the diagnosis of AL amyloidosis. The patient received symptomatic treatment with diuretics and therapeutic anticoagulation, followed by a specific regimen comprising cyclophosphamide, bortezomib, and dexamethasone. The patient's hematologic response was characterized by normalization of free light chain levels, while the cardiac response included reductions in B-type natriuretic peptide (BNP) and troponin levels, alongside improved global longitudinal strain.
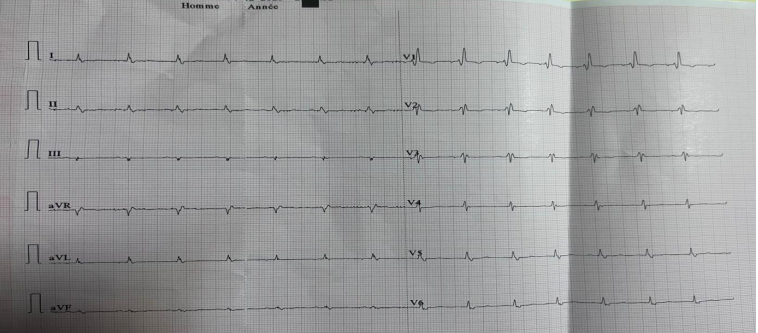
Figure 1. diffuse low voltage with right bundle branch block
Discussion
Cardiac amyloidoses comprise a diverse group of diseases characterized by extracellular accumulation of amyloid fibrillar proteins that progressively infiltrate tissues, leading to dysfunction. Presently, 36 amyloid proteins are known, with the most frequent cardiac forms being AL amyloidosis and mutated or senile TTR amyloidosis (ATTR) [4].
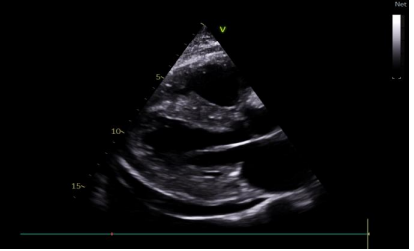
Figure 2. symmetrical hypertrophic cardiomyopathy affecting the septum and posterior wall with Pericardial Effusion
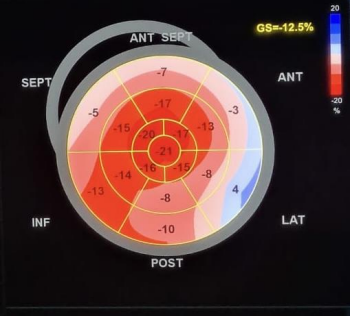
Figure 3. Global longitudinal strain with rosette-like appearance
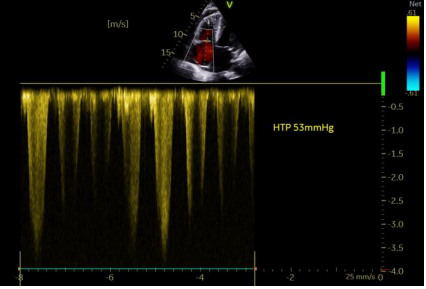
Figure 4. Pulmonary hypertension by Tricuspid regurgitation jet
AL Amyloidosis Pathophysiology:
AL amyloidosis arises from the secretion of immunoglobulin light chains by plasma cell clones, forming amyloid deposits that impair various organ tissues. This condition often coexists with monoclonal gammopathies and hematologic malignancies, sometimes serving as their revealing mode. Understanding these pathophysiologies is crucial for comprehending drug mechanisms [5].
Diagnostic Considerations:
Clinicians should consider cardiac and extracardiac signs, interpreting ECGs in the clinical and echocardiographic context. A crucial diagnostic step is evaluating the mismatch between electrical microvoltage and echocardiographic hypertrophy. Immunologic assessments and bone scintigraphy aid in differentiating between AL and ATTR amyloidoses, with histological proof required for AL amyloidosis before initiating chemotherapy [6].
ECG Findings:
Microvoltage, though present in only 60% of AL amyloidoses, serves as a prognostic factor indicating advanced disease. Conduction abnormalities, such as branch blocks and first-degree atrioventricular block, are common, justifying close monitoring. Red flags include relative microvoltage, pseudo-Q waves, unexplained conduction disturbances, and atrial fibrillation [7].
Echocardiographic Features:
Amyloid deposits can affect not only the left ventricle but also the right ventricle, atria, and interatrial septum. Atrial septal hypertrophy, coupled with increased myocardial echogenicity, is highly specific for amyloidosis. Global longitudinal strain (GLS) of the left ventricle serves as an early diagnostic marker and a major prognostic factor [8].
Laboratory Assessments:
Laboratory investigations, especially comprehensive immunologic assessments, are essential to rule out AL amyloidosis promptly. The importance of three essential laboratory examinations, serum protein electrophoresis, immunofixation of serum and urine proteins (Bence Jones), and serum free light chain assay with a K/L ratio, cannot be overstated. Cardiac biomarkers play a crucial role in early suspicion, with persistent elevation in troponin and BNP levels providing both diagnostic and prognostic value. While laboratory tests aid in early detection, confirmation through biopsy remains indispensable, especially in cases of AL amyloidosis [9].
Biopsy for Diagnosis Confirmation:
Extracardiac biopsy sites include subcutaneous fat or rectum, with less frequent involvement of solid organs such as the heart, liver, or kidney. Two separate specimens are necessary for Congo red staining and yellow-green birefringence in polarized light, as well as protein typing through immunohistochemistry/spectrometry [10].
Symptomatic Treatment and Thromboprophylaxis:
Symptomatic treatment centers around diuretics and sodium restriction, tailored to congestion levels [11]. Amyloidosis carries a high thrombotic risk due to the nephrotic syndrome associated with AL amyloidosis, necessitating formal anticoagulation. Additionally, the dissociation between atrial mechanical and electrical activity makes atrial thrombus formation a risk even in sinus rhythm. Thromboembolic events should be screened for and anticoagulated irrespective of the CHA2DS2-VASc score [12]. Beta-blockers and bradycardic agents are not recommended, as a higher heart rate is crucial for maintaining cardiac output due to the stiffness of the ventricular wall [13].
Management of Atrial Fibrillation:
Amiodarone is the preferred choice for maintaining sinus rhythm in atrial fibrillation. Digoxin is discouraged due to potential toxic effects on atrial rhythm disturbances [14].
Treatment of AL Amyloidosis:
AL amyloidosis is a hematologic emergency, treated with chemotherapy combining cyclophosphamide and bortezomib, along with immunotherapy using an anti-CD38 monoclonal antibody targeting plasma cells. Monitoring the hematologic and cardiac response is essential. Hematologic response is assessed through serum free light chain measurement, with complete, partial, or no response criteria [15]. Cardiac response evaluation requires a more extended period, necessitating closer clinical and laboratory surveillance. Monthly follow-ups are recommended initially, transitioning to quarterly assessments after the initial treatment phase, including ECG and echocardiography every six months and annual Holter monitoring [2]. Regular monitoring ensures comprehensive evaluation and adaptation of the therapeutic strategy, optimizing patient outcomes in the management of cardiac amyloidosis [16].
Conclusion
This case underscores the complexity of AL amyloidosis, emphasizing the intricate interplay of cardiac, renal, and hematologic manifestations. Early diagnosis and a tailored therapeutic approach contributed to positive outcomes in both hematologic and cardiac parameters. This case contributes to the growing body of literature on the multifaceted nature of amyloidosis and highlights the importance of a multidisciplinary approach in its management.
Declarations Consent for publication
Written informed consent was obtained from the patients for publication of this case report and any accompanying images. Availability of data and material: All data analysed during this study are included in this published article.
Competing interests: The authors declare that they have no competing interests.
Author contributions: Zahri Soukaina: writing paper, Mehdi Belhakim: study concept literature, Wakil Nouhaila: interpretation and analysis, Rachida Habbal: Interpretation and analysis
References
- Dubrey SW, Hawkins PN, Falk RH. (2011) Amyloid diseases of the heart: assessment, diagnosis, and referral. Heart. 97(1): 75‑84. [PubMed.]
- Garcia-Pavia P, Rapezzi C, Adler Y, Arad M, Basso C, et al. (2021) Diagnosis and treatment of cardiac amyloidosis: a position statement of the ESC Working Group on Myocardial and Pericardial Diseases. European heart journal. 42(16): 1554‑1568. [PubMed.]
- Oerlemans MIFJ, Rutten KHG, Minnema MC, Raymakers RAP, Asselbergs FW, et al. (2019) Cardiac amyloidosis: the need for early diagnosis. Neth Heart J. 27(11): 525‑536. [PubMed.]
- De Marneffe N, Dulgheru R, Ancion A, Moonen M, Lancellotti P. (2022) Cardiac amyloidosis: a review of the literature. Acta Cardiologica. 77(8): 683‑692. [PubMed.]
- Siddiqi OK, Ruberg FL. (2018) Cardiac amyloidosis: an update on pathophysiology, diagnosis, and treatment. Trends in cardiovascular medicine. 28(1): 10‑21. [PubMed.]
- Wechalekar AD, Fontana M, Quarta CC, Liedtke M. (2022) AL Amyloidosis for Cardiologists. JACC: CardioOncology. 4(4): 427‑441. [PubMed.]
- Mohty D, Damy T, Cosnay P, Echahidi N, Casset-Senon D, et al. (2013) Cardiac amyloidosis: updates in diagnosis and management. Archives of cardiovascular diseases. 106(10): 528‑540. [PubMed.]
- Salinaro F, Meier-Ewert HK, Miller EJ, Pandey S, Sanchorawala V, et al. (2017) Longitudinal systolic strain, cardiac function improvement, and survival following treatment of light-chain (AL) cardiac amyloidosis. European Heart Journal-Cardiovascular Imaging. 18(9): 1057‑1064. [PubMed.]
- Vergaro G, Aimo A, Barison A, Genovesi D, Buda G, et al. (2020) Keys to early diagnosis of cardiac amyloidosis: red flags from clinical, laboratory and imaging findings. European Journal of Preventive Cardiology. 27(17): 1806‑1815. [PubMed.]
- Kapoor P, Thenappan T, Singh E, Kumar S, Greipp PR. (2011) Cardiac amyloidosis: a practical approach to diagnosis and management. The American journal of medicine. 124(11): 1006‑1015. [PubMed.]
- Grogan M, Dispenzieri A. (2015) Natural history and therapy of AL cardiac amyloidosis. Heart Fail Rev. 20(2): 155‑1562. [PubMed.]
- Bukhari S, Khan SZ, Bashir Z. (2023) Atrial fibrillation, thromboembolic risk, and anticoagulation in cardiac amyloidosis: a review. Journal of Cardiac Failure. 29(1): 76‑86. [PubMed.]
- Adam RD, Coriu D, Jercan A, Bădeliţă S, Popescu BA, et al. (2021) Progress and challenges in the treatment of cardiac amyloidosis: a review of the literature. ESC Heart Failure. Août. 8(4): 2380‑2396. [PubMed.]
- Cappelli F, Tini G, Russo D, Emdin M, Del Franco A, et al. (2021) Arterial thrombo-embolic events in cardiac amyloidosis: a look beyond atrial fibrillation. Amyloid. 28(1): 12‑18. [PubMed.]
- Bianchi G, Zhang Y, Comenzo RL. (2021) AL amyloidosis: current chemotherapy and immune therapy treatment strategies: JACC: CardioOncology state-of-the-art review. JACC: CardioOncology. 3(4): 467‑487. [PubMed.]
- Banypersad SM, Moon JC, Whelan C, Hawkins PN, Wechalekar AD. (2012) Updates in Cardiac Amyloidosis: A Review. JAHA. 1(2): e000364. [PubMed.]