>Corresponding Author : Iqra Ayaz Awan
>Article Type : Case Report
>Volume : 2 | Issue : 6
>Received Date : 26 Sep, 2022
>Accepted Date : 06 Oct, 2022
>Published Date : 10 Oct, 2022
>DOI : https://doi.org/10.54289/JCRMH2200131
>Citation : Awan IA. (2022) Bart’s Syndrome: A Case Report. J Case Rep Med Hist 2(6): doi https://doi.org/10.54289/JCRMH2200131
>Copyright : © 2022 Awan IA. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Case Report | Open Access | Full Text
Consultant General Paediatrics, Shaafi International Hospital
*Corresponding author: Iqra Ayaz Awan, Consultant General Paediatrics, Shaafi International Hospital
Abstract
Bart's syndrome (BS) is a genetic disorder characterized by the absence of localized skin (present at birth), epidermolysis bullosa (EB) and ungueal changes [1]. Clinically Bart's syndrome is associated with epidermolysis bullosa (EB). It may be associated with any type of EB but is mostly reported with dominant dystrophic form. Diagnosis is obvious clinically but requires ultrastructural microscopy. Treatment suffices to conservative measures [1].
Abbreviations: BS: Bart's Syndrome, EB: Epidermolysis Bullosa, OFC: Occipitofrontal Circumference, CLAS: Congenital Localized Absence of Skin, ACC: Aplasia Cutis Congenita
Introduction
Bart syndrome is a rare neonatal pathology combining congenital skin aplasia affecting the extremities and exceptionally described congenital epidermolysis bullosa [2]. Bart's syndrome is an autosomal dominant inheritance caused by mutations in the type VII collagen gene on chromosome 3p3. It was first described in a large family almost half a century ago [3]. Bart et al reported the case of 26 members of a family who had congenital absence of skin in the lower limbs, mucous blisters, and absence or deformity of the nails [4]. Any part of skin can be involved but disease tends to occur more on extremities and parts exposed to friction and trauma [1].
The purpose of reporting this case is it being rare and how our timely conservative management of skin condition along with nutrition led to the favorable outcome of this non curable congenital disorder, thus avoiding the need for complex surgical management like auto or allografts.
Case Report
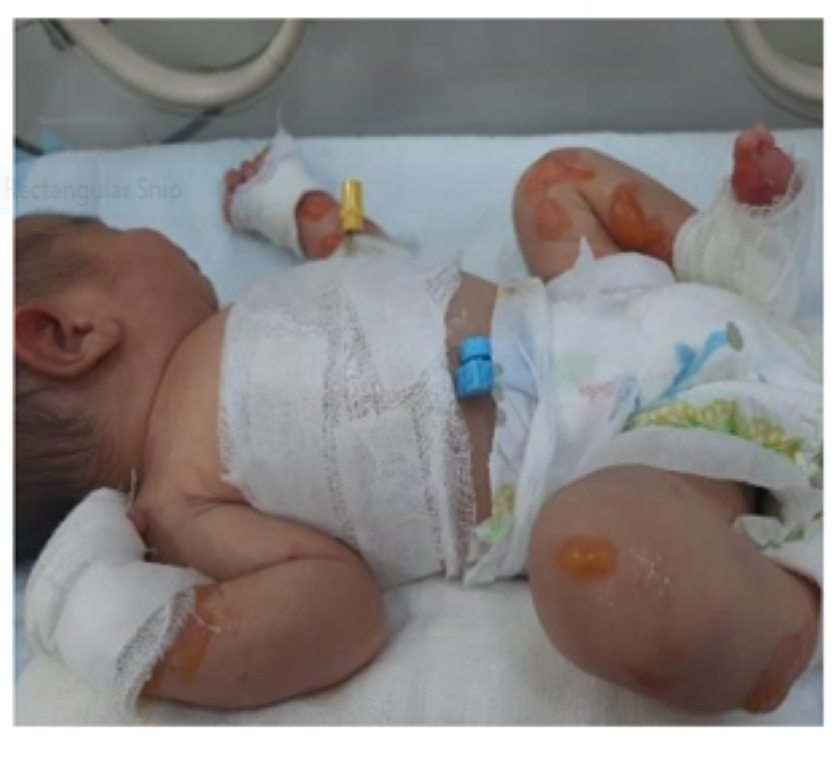
A male infant born at 37 weeks with circumferential absence of all layers of skin on feet along with multiple areas of blisters and skin erosions all over the body to a 21years old G2 P1 mother via uneventful spontaneous vaginal delivery; other sibling is a healthy 2 years old female. Pregnancy was uncomplicated with normal anomaly scan done at 20th week of gestation. Parents were a consanguineous healthy couple with no personal or family history of skin disorders. Mother had no history of drug intake or radiation exposure during pregnancy.
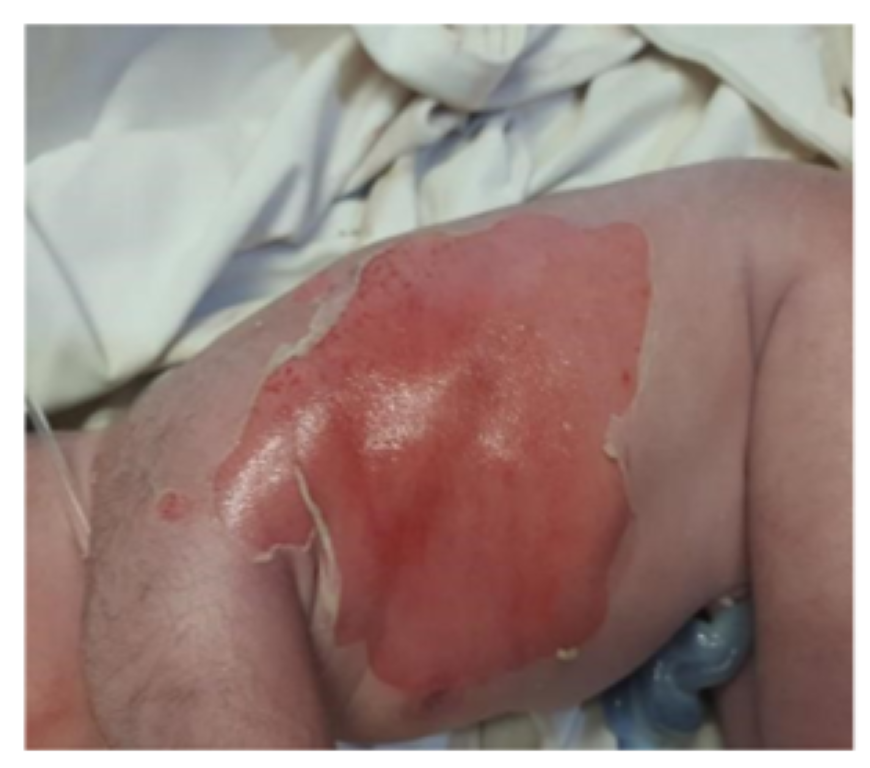
The baby was born with APGAR score of 8 and 9 at 1 and 5 minutes of life respectively. On clinical examination vitals, weight, occipitofrontal circumference (OFC), length and systemic examination were completely normal. However, head to toe examination revealed skin defects on feet with multiple areas of skin erosions over scalp, trunk, nape of neck, cheeks and left. Few blisters could be appreciated around elbow and knee joint area which were suggestive of epidermolysis bullosa. There were no oral erosions.
Circumferential defect of all layers of skin around the ankle extending proximally to just behind the ankle joint and distally onto the dorsal aspect of the foot covering the metatarsals up till the metatarsophalangeal joints having well demarcated borders. Underlying vascular network and muscles could be appreciated with only covering being a clear glistening membrane. Toe nails were normal.
Extent of involvement:
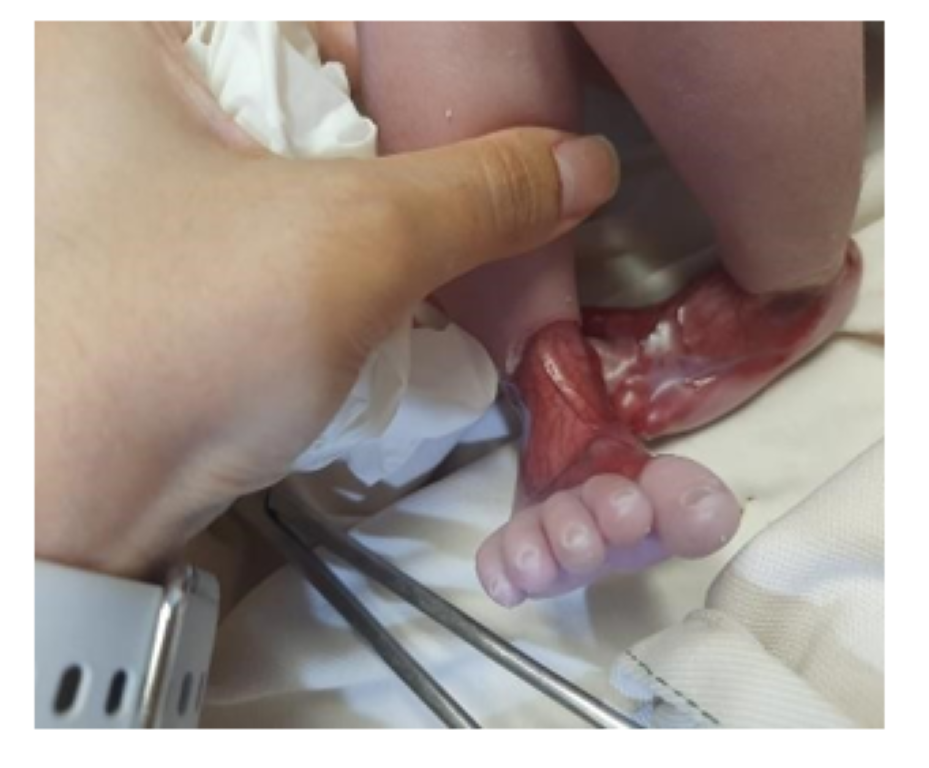
RIGHT FOOT: Circumferential defect of all layers of skin around the ankle extending proximally to just behind the ankle joint and distally onto the dorsal aspect of the foot covering the metatarsals up till the metatarsophalangeal joints having well demarcated borders. Underlying vascular network and muscles could be appreciated with only covering being a clear glistening membrane. Toe nails were normal.
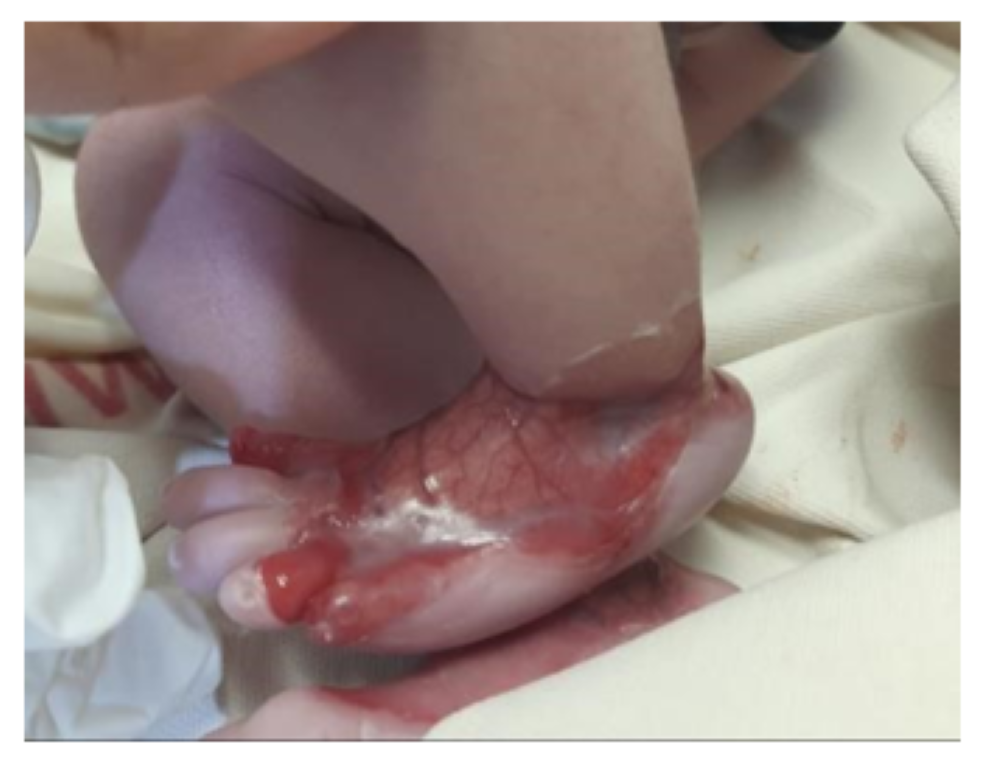
LEFT FOOT: Circumferential absence of all layers of skin creating a defect starting proximally from around the ankle joint and extending distally onto the dorsum of foot and 1st, 5th and proximal part of the 4th toe with clearly defined margins. Underlying vascular structures were clearly visible and nails of 1st and 5th toes were dystrophic.
There were no other associated physical abnormalities. Baby was admitted to NICU for further work up and management. He was taking adequate feeds and passing stools and had no episode of vomiting or abdominal distention.
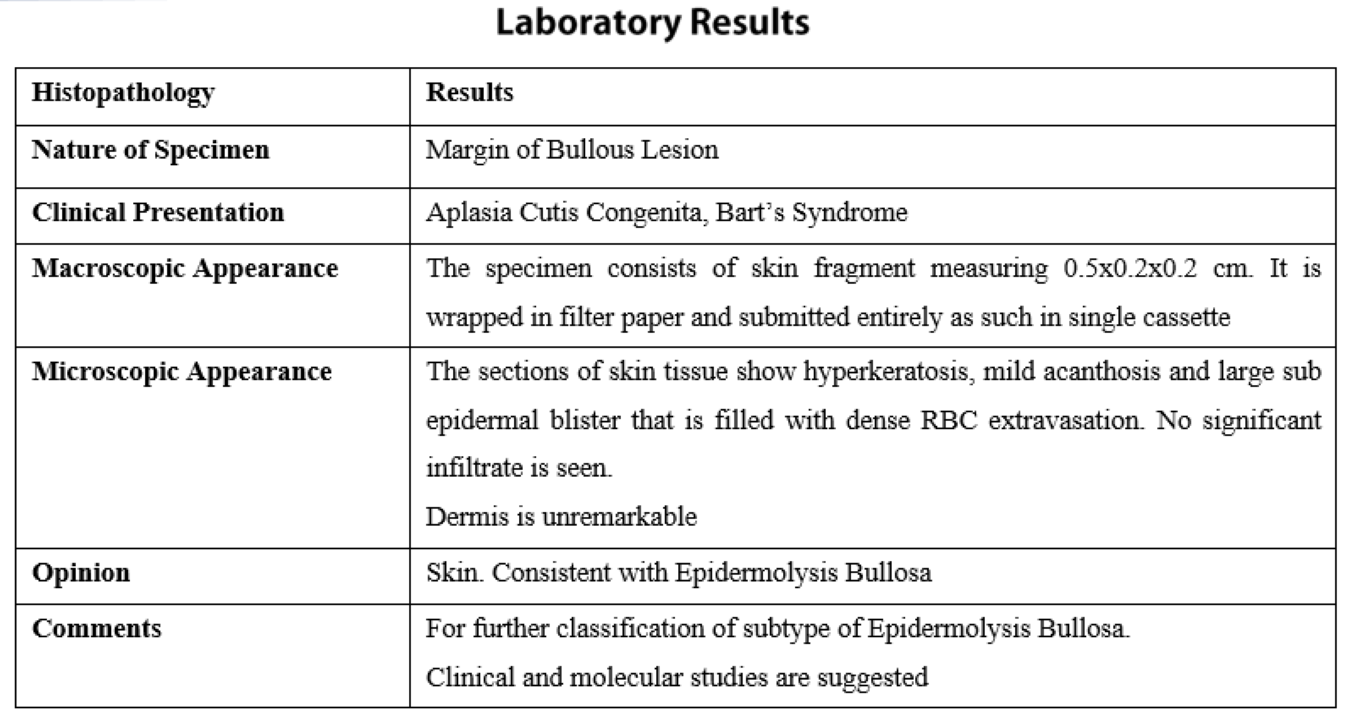
Results of lab investigations which included Complete Blood Count, C-Reactive Protein, blood Culture and sensitivity, serum electrolytes, Liver Function Tests and Renal Function Tests were within normal limits. Skin biopsy was taken from a fresh blister on right leg along with skin from adjacent normal area and sent for histopathology. Management included systemic antibiotics (cefotaxime, amikacin). Skin lesions and wounds were managed conservatively with dressings and paraffin gauze.
Feet wounds /ulcers were dressed for twice daily for 5days when they were first washed with normal saline and povidine, followed by application of Fucidin cream which were then covered with chlorhexidine plus paraffin dressing (bactigras) and loose sterile dressings. Rest of the minor skin erosions were cleaned with normal saline followed by Fucidin cream application and were then left uncovered. Small blisters /bullae were left intact (unpopped).
Baby was sent home after 5 days with all the necessary home care instructions in detail. Parents were advised to bathe the baby daily with normal saline and skin erosions to be covered with sterile paraffin gauze. Foot dressings were advised to continue daily for 9 days. Follow up was advised at 2 weeks of life. Follow up was done at 2 weeks of life.
Direct immunofluorescence and genetic testing was not done due to unaffordability and unavailability respectively.
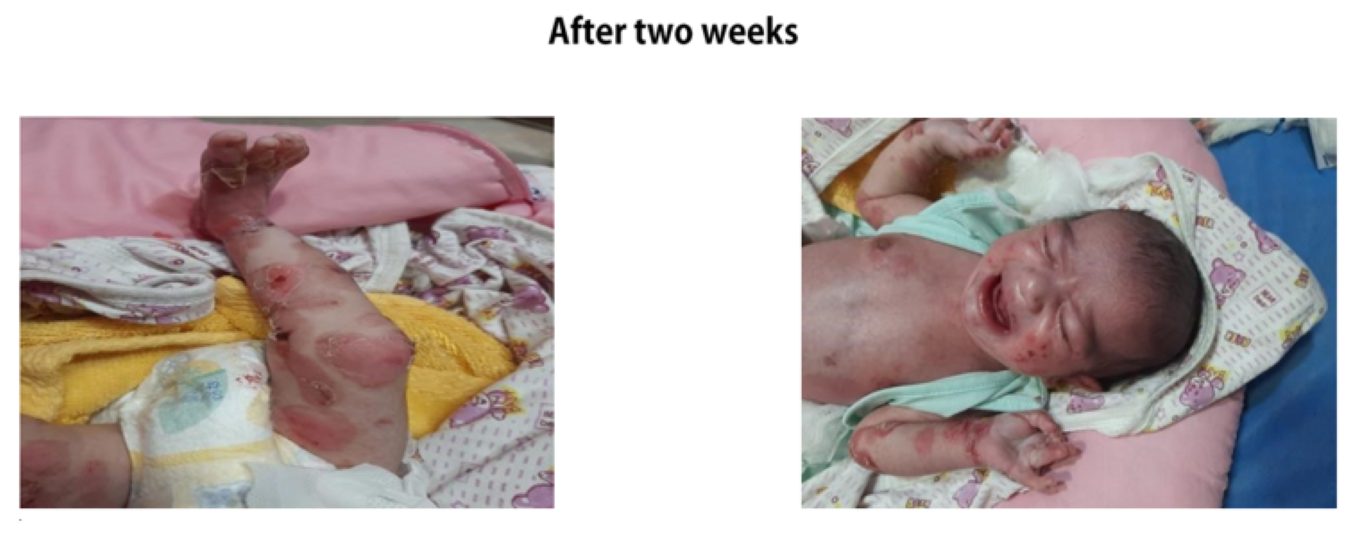
Discussion
Bart syndrome is considered exceedingly rare genetic disorder. It presents with the classic triad of congenital localized absence of skin over both lower legs, blistering of the skin, and nail dystrophy. The inheritance pattern of Bart syndrome appears to be autosomal dominant but sporadic cases have been reported [2]. Congenital localized absence of skin (CLAS) occurs in association with all three major types of inherited epidermal diseases and merits retaining its status as a unique clinical entity [1].
The etiopathogenesis of aplasia cutis congenita (ACC) remains unclear. The condition encompasses a heterogeneous group of disorders with or without associated abnormal physical findings, malformation patterns, or genetic diseases [5]. Bart's syndrome may also be akin to other anomalies such as pyloric atresia, flattened nose, broad nasal root, and wideset eyes [6,7]. Some authors propound it to be caused by intrauterine physical trauma, while others debate that the affected area follows Blaschko's lines [4,8].
Cases of aplasia cutis congenita (ACC) secondary to drugs such as methimazole and diclofenac sodium have already been reported. Desloratadine use in pregnancy is associated with hypospadias. In another case described in literature, ACC occurred due to montelukast use [7]. The first case of ACC on the leg without association with epidermolysis bullosa was reported in Korea which after conservative treatment with prophylactic topical antibiotics and wet dressing healed with atrophic scar formation and after 2 years there was little scar formation and no functional impairment of the leg [5,9].
Antenatal ultrasound may show polyhydramnios and gastric dilatation as manifestations of upper gastrointestinal obstruction if associated with pyloric atresia [10]. The management of BS is conservative and involves close follow-up for serious complications, such as hemorrhage, infection, hypothermia, and hypoglycemia [11]. The primary approach towards treatment is to promote the epithelialization and healing of skin lesions conservatively without complications which are reported to be infections (secondary sepsis, localized superinfection), hemorrhage due to skin fragility, dehydration and hydro electrolytic disorders [3]. Restorative surgery may be needed if conservative management fails
In the present case, the aplasia manifested bilaterally in the lower limbs. The management of this syndrome involves conservative measures for treatment of wounds. The administration of systemic antibiotics is not routine but can be used if an infection is suspected [4,12].
Conclusion
To achieve the best outcome, care and diagnosis must be accomplished as early as possible [13]. Bart syndrome is an odd congenital connection of three distinct skin symptoms-ACC, EBC, and ungeal involvement-whose etiopathogeny is still poorly understood. The prognosis is directly influenced by the management, which is difficult even though the clinical diagnosis is straightforward and aims to treat the skin lesions as effectively as possible while preventing surgery as well as subsequent problems [3].
References
- Bajaj DR, Qureshi A. (2008) Bart’s Syndrome: A Case Report. Journal Of Pakistan of Dermatologists. 18: 113-115. [Ref.]
- Alfayez Y, Alsharif S, Santli A. (2017) A Case of Aplasia Cutis Congenita Type VI: Bart Syndrome. Case Rep Dermatol. 9: 112-118. [PubMed.]
- Cissouma A, Kassogue D, Sylla M, Demebele G, Traore SA, et al. (2021) Bart’s Syndrome: A Neonatal Observation About a Case Report. Open Journal of Pediatrics. 11: 406-412. [Ref.]
- Teles RA. (2016) Bart Syndrome: A Case Report, Residencia Pediatrica. 6(2): 94-97. [Ref.]
- Lee JS, Yun SJ, Lee JB, Kim SJ, Won YH, et al. (2008) A Case of Aplasia Cutis Congenita, Type VII. Annals Of Dermatology. 20(2): 70-73. [PubMed.]
- Cicekci F. (2022) Anesthesia Management in Bart’s Syndrome: A Case Report. Acta Medica Alanya. 6(1): 114-117. [Ref.]
- Rajpal A, Mishra R, Hajirnis K, Shah M, Nagpur N. (2008) Bart’s Syndrome. Indian J Dermatol. 53: 88-90. [Ref.]
- Sharma D, Lamba S, Maheshwari A, Shastri S. (2016) A Neonate with Aplasia Cutis Congentia Type VI with Junctional Epidermolysis Bullosa: A Very Rare Condition. Medical Journal of Islamic World Academy of Science. 24(3): 96-98. [Ref.]
- Satyanarayana VVV. (2015) Bart’s Syndrome: A Case Report. SSRG International Journal of Medical Science. 2(9). [Ref.]
- Basting O, Ozdemir A, Korkut S, Korkmaz L, Halis H, et al. (2017) A Rare Association: Bart’s Syndrome and Congenital Adrenal Hyperplasia Associated with Drugs Embryopathy? J Clin Neonatal. 6: 250-253. [Ref.]
- Kulai F, Bas AY, Kale Y, Celik IH, Demirel N, et al. (2015) Type VI Aplasia Cutis Congenita: Bart’s Syndrome. Case Reports in Dermatological Medicine Article. ID 549825. [Ref.]
- Mead A, Alem Y, Sheikh OA, Hussein LI. (2021) A Case Report of Aplasia Cutis Congenita Type VI: Bart’s Syndrome. International Journal Advanced Research. 9(12): 251-254. [Ref.]
- Al-Gburi S, Namuq Z. (2022) Twin Neonates with Bart’s Syndrome. Cureus. 14(1): E21363. [Ref.]